Introducción a los Programas de Cribado Neonatal
Los programas de cribado neonatal (PCN) son esenciales para la prevención en Salud Pública y están reconocidos en los diferentes sistemas sanitarios. Aunque su diseño y recursos pueden variar entre las distintas comunidades, su objetivo fundamental es la detección temprana de enfermedades en recién nacidos (RN).
En España, el cribado neonatal se realiza mediante una muestra de sangre impregnada en papel para detectar al menos ocho entidades, entre las que se incluyen el hipotiroidismo congénito (HC), la fenilcetonuria (PKU), diversas deficiencias enzimáticas, la fibrosis quística (FQ) y la anemia de células falciformes (AF).
Los inicios del cribado neonatal se remontan a la década de 1960 con la prueba bacteriológica de Guthrie para detectar la fenilalanina. Con el avance de las técnicas analíticas, especialmente la espectrometría de masas en tándem (MS/MS), se ha expandido el cribado metabólico, permitiendo la detección de un mayor número de enfermedades.
Propósito y Principios del Cribado Neonatal
El propósito de los análisis de cribado neonatal es identificar a los recién nacidos presuntamente positivos para un trastorno específico, minimizando la tasa de falsos positivos. Esto se logra estableciendo puntos de corte arbitrarios para los analitos o marcadores cuantificados.
Es crucial entender que los PCN no son procedimientos de diagnóstico definitivo. Identifican grupos de alto riesgo que requieren estudios posteriores. Por ello, los PCN deben integrarse con unidades clínicas y de laboratorio especializadas para el diagnóstico y tratamiento de las enfermedades detectadas.
El principal beneficio de un PCN es la prevención de discapacidades asociadas a la enfermedad. La estrategia de planificación de estos programas debe asegurar una cobertura del 100% de los RN y el tratamiento temprano del 100% de los casos detectados.
Criterios para la Inclusión de Trastornos en el Cribado Masivo
Clásicamente, se definieron criterios para incluir un trastorno en el cribado masivo:
- La enfermedad debe cursar con morbilidad significativa (retraso mental) o mortalidad si no se diagnostica en el período neonatal.
- Debe existir un tratamiento eficaz.
- La frecuencia de la enfermedad debe ser relativamente elevada (al menos, 1 de cada 10.000-15.000 RN).
- Debe existir un método analítico de cribado rápido, fiable y de bajo coste.
Con la incorporación de la tecnología MS/MS, la prevalencia evaluada puede ser la suma de la prevalencia de cada enfermedad detectada, permitiendo considerar el cribado de enfermedades con muy baja prevalencia (< 1:50.000 RN).
Recomendaciones y Estrategias de Cribado
En 2005, la Academia Americana de Pediatría (AAP) y la Asociación Americana de Genética Médica (ACMG) emitieron recomendaciones para el cribado de trastornos en EE.UU. Esto ha llevado a que en algunas regiones se criben más de 50 condiciones genéticas, a pesar de la necesidad de familiarizar a los profesionales de Atención Primaria con estas patologías.
Como norma general, se recomienda una extracción única de sangre capilar a partir de las 48 horas de vida del RN, tras una ingesta de 24 horas. La extracción debe ser realizada exclusivamente por personal sanitario, utilizando la punción del talón como procedimiento habitual.
Para minimizar el dolor, se pueden emplear medidas como la lactancia materna, soluciones de glucosa o chupetes con sacarosa. La punción debe realizarse con lanceta estéril en la porción medial o lateral de la superficie plantar del talón para evitar daños osteo-articulares. El pie debe masajearse previamente para aumentar el flujo sanguíneo y desinfectarse con alcohol de 70º.

Tras la punción, se limpia la primera gota de sangre, se deja formar una nueva gota grande y se aplica sobre el papel absorbente, llenando el círculo de forma uniforme. La muestra debe secarse en una superficie limpia, seca y no absorbente, a temperatura ambiente y evitando la luz solar directa, antes de enviarse al laboratorio lo antes posible (preferiblemente dentro de las 24-48 horas siguientes a la extracción).
Existen también estudios de cribado a partir de muestras de orina utilizando técnicas MS/MS o cromatografía de gases y espectrometría de masas (GC/MS), que permiten la confirmación diagnóstica, complementar la información de la muestra de sangre o ampliar horizontes a nuevos diagnósticos. Algunas regiones de Canadá y países asiáticos han iniciado PCN en muestras de orina.
Consideraciones Especiales en la Toma de Muestras
Se deben tener en cuenta situaciones clínicas particulares:
- RN con patología grave al nacimiento: se recomienda tomar una muestra al ingreso (si es posible antes de tratamiento antibiótico o alimentación parenteral) y repetirla a las 48-72 horas.
- Transfusiones previas a la toma de muestra: obligan a realizar una toma de muestra a las 48-72 horas de la transfusión y repetirla a los 120 días.
- RN alimentados con alimentación parenteral: se debe realizar una toma de muestra a las 48-72 horas después de iniciar la alimentación enteral, independientemente de la edad del niño.
Enfermedades Incluidas en el Cribado Metabólico Neonatal
Hipotiroidismo Congénito (HC)
El HC tiene una importancia extraordinaria debido a su potencial repercusión en el desarrollo intelectual. Las hormonas tiroideas son imprescindibles para el desarrollo cerebral, siendo esta la causa de retraso mental prevenible más frecuente. Su frecuencia (1/3.500 RN vivos) justifica el programa de cribado neonatal.
El diagnóstico precoz con inicio de tratamiento en el primer mes de vida se asocia a coeficientes de inteligencia normales y desarrollo satisfactorio. Existen formas transitorias de HC debidas a prematuridad, uso de compuestos yodados o consumo materno de ciertos medicamentos.
El cribado sistemático se basa en la determinación del nivel de TSH en sangre del talón. Un nivel de TSH elevado (≥ 10µUI/ml) indica hipotiroidismo primario. La confirmación se realiza mediante la medida de T4 libre sérica, que habitualmente está descendida. Pueden existir alteraciones intermedias como hipertirotropinemia o hipotiroxinemia.
Fenilcetonuria (PKU)
La PKU es una enfermedad del metabolismo de la fenilalanina (Phe) con herencia autosómica recesiva. Su prevalencia oscila entre 1/12.000-1/18.000 RN. El cribado se realiza midiendo por MS/MS la Phe y tirosina (Tyr) en sangre extraída a las 48-72 horas de vida, impregnada en papel absorbente. La alta sensibilidad de esta tecnología permite adelantar la toma de muestras.
Trastornos de la Beta-Oxidación Mitocondrial
Estos trastornos de los ácidos grasos, como la MCADD o LCHADD, se transmiten de forma autosómica recesiva. Su prevalencia es variable (1/9.000 a 1/100.000 RN). Se produce un acúmulo de ácidos grasos y sus derivados, lo que genera problemas fisiopatológicos, defecto de energía o acumulación de moléculas tóxicas, manifestándose en situaciones de descompensación metabólica.
Aciduria Glutárica Tipo I (AG-1)
La AG-1 es una enfermedad del metabolismo de los ácidos orgánicos causada por un defecto en la enzima GCDH, con herencia autosómica recesiva y una prevalencia en España de 1/85.000 RN. La enzima metaboliza los aminoácidos lisina (Lys) y triptófano (Trp). Su deficiencia provoca la acumulación de compuestos tóxicos para el sistema nervioso.
El bebé no presenta problemas al nacer, pero la acumulación de derivados tóxicos comienza con la alimentación. Procesos infecciosos, fiebre o ayuno prolongado pueden desencadenar crisis encefalopáticas.
Deficiencia de Biotinidasa (BTD)
El BTD es una forma de aparición tardía del déficit múltiple de carboxilasas. Si no se trata, se caracteriza por convulsiones, dificultad para respirar, hipotonía, erupciones cutáneas, alopecia, pérdida de audición y retraso en el desarrollo. La prevalencia clínica se estima en 1/61.000 RN, con una frecuencia de portadores de aproximadamente 1/120.
Los síntomas suelen aparecer en los primeros meses de vida, pero también pueden manifestarse posteriormente. Los individuos con déficit profundo sin tratar presentan hallazgos clínicos variables, acidosis cetoláctica, acidemia orgánica e hiperamonemia leve. Los individuos con déficit parcial pueden ser asintomáticos, pero desarrollar síntomas durante periodos de estrés. El tratamiento principal es la suplementación con biotina oral, que mejora los síntomas y previene su aparición. El tratamiento debe mantenerse de por vida, y los rasgos establecidos como atrofia óptica o pérdida de audición pueden no ser reversibles.
Manejo de Resultados Alterados en el Cribado Neonatal
Los resultados alterados en el cribado neonatal requieren un manejo específico:
- Resultados alterados, altamente sugestivos de una alteración metabólica grave: Contactar lo antes posible con la familia mediante llamada telefónica por un profesional con experiencia en el manejo de enfermedades metabólicas. Se debe explicar a los padres el significado del resultado y los pasos a seguir para confirmar o excluir el diagnóstico.
- Resultados fuera del rango normal, pero no tan alterados como para indicar una enfermedad grave: Estos casos sospechosos requieren una repetición del test. La solicitud de nueva muestra se realiza generalmente por carta del laboratorio de cribado, explicando la necesidad de una muestra adicional para descartar un resultado falso positivo. Se aconseja a los padres acudir a su pediatra para la repetición del test y recibir información adicional.
Fibrosis Quística (FQ) en el Cribado Neonatal
La Fibrosis Quística (FQ) es una enfermedad crónica, potencialmente grave y progresiva, que afecta principalmente a los pulmones y al sistema digestivo. Su inclusión en el cribado neonatal permite la detección precoz antes de la aparición de la sintomatología clínica, mejorando el pronóstico del paciente.
El cribado de FQ en recién nacidos se realiza mediante un examen de sangre que busca niveles elevados de tripsinógeno inmunorreactivo (TIR), una proteína producida por el páncreas relacionada con la FQ. La muestra se extrae del talón del bebé, se recoge en papel de filtro y se envía a laboratorio.

Un resultado anormal (positivo) en el cribado sugiere que el niño puede tener FQ, pero no es un diagnóstico definitivo. Se requieren pruebas adicionales para confirmar o descartar la enfermedad. La prueba de cloruro en sudor es el estándar diagnóstico para FQ.
El diagnóstico temprano y el inicio del tratamiento a edad temprana pueden mejorar la nutrición, el crecimiento y el funcionamiento pulmonar de los niños con FQ. Algunos estados incluyen este examen dentro de las pruebas de detección de rutina del recién nacido.
Diagnóstico y Confirmación de la Fibrosis Quística
Si la prueba del talón da positivo en fibrosis quística, la familia se derivará a una unidad de referencia para cribado neonatal de fibrosis quística. Las pruebas de confirmación incluyen el análisis molecular para identificar mutaciones en el gen CFTR, el test del sudor y la evaluación de la función exocrina.
El test del sudor analiza la conductividad y concentración de cloro en una muestra de sudor obtenida tras estimular la piel con pilocarpina. Un alto nivel de sal en el sudor es una señal de la enfermedad.
La identificación de dos variantes de genes que causan FQ en pacientes con un resultado positivo de cribado neonatal o características clínicas típicas también contribuye al diagnóstico.
Mecanismos Moleculares y Genética de la Fibrosis Quística
La FQ es una enfermedad genética causada por disfunción de la proteína reguladora de la conductancia transmembrana de la fibrosis quística (CFTR). El gen CFTR, localizado en el cromosoma 7, codifica un canal de cloruro regulado por cAMP que controla el transporte de cloruro, sodio y bicarbonato a través de las membranas epiteliales.
La enfermedad se manifiesta en personas homocigotas para variantes patogénicas bialélicas. La variante génica más común es F508del, presente en aproximadamente el 85% de los alelos con FQ. Se han identificado más de 2.000 variantes menos comunes.
Las variantes de CFTR se han clasificado en 6 clases según su efecto en la función o procesamiento de la proteína. Las variantes de clase I, II o III producen escasa o ninguna función de CFTR, mientras que las de clase IV, V o VI se asocian con función residual. No obstante, la relación entre variantes específicas y la manifestación de la enfermedad no es estricta, siendo las pruebas clínicas una mejor guía pronóstica.
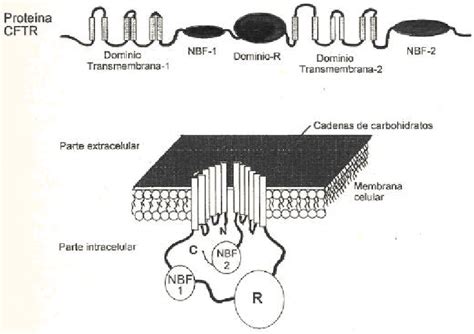
Fisiopatología de la Fibrosis Quística
La FQ afecta a casi todas las glándulas exocrinas, con distribución y gravedad variables.
Aparato Respiratorio
En la mayoría de los pacientes, la enfermedad pulmonar se manifiesta desde la lactancia o infancia temprana. El taponamiento por moco y la infección bacteriana crónica, junto con una respuesta inflamatoria pronunciada, lesionan las vías respiratorias, provocando bronquiectasias e insuficiencia respiratoria. La evolución clínica se caracteriza por exacerbaciones episódicas, infección y declinación progresiva de la función pulmonar.
El daño pulmonar se desencadena por la obstrucción difusa de las pequeñas vías respiratorias por secreciones mucosas anormalmente espesas. La inflamación crónica también contribuye a la lesión pulmonar. La hiperreactividad bronquial se observa en aproximadamente el 40% de los pacientes.
En enfermedad pulmonar avanzada, la hipoxemia crónica induce hipertrofia muscular de las arterias pulmonares, hipertensión pulmonar e hipertrofia ventricular derecha. Los pulmones suelen estar colonizados por bacterias patógenas como Staphylococcus aureus y Pseudomonas aeruginosa, así como otros patógenos como SARM, el complejo Burkholderia cepacia y micobacterias no tuberculosas.
Aparato Digestivo
El páncreas, el intestino y el sistema hepatobiliar suelen estar afectados. En aproximadamente el 85% de los pacientes, hay compromiso de la función exocrina del páncreas, lo que provoca malabsorción de grasas, vitaminas liposolubles y proteínas, resultando en esteatorrea, desnutrición y crecimiento insuficiente en niños.
La disfunción pancreática endocrina puede llevar a intolerancia a la glucosa o diabetes mellitus. La enfermedad hepática relacionada con la FQ ocurre en aproximadamente el 30% de los pacientes, pudiendo progresar a cirrosis irreversible.
En recién nacidos, las secreciones intestinales anormalmente viscosas pueden causar íleo meconial. Niños mayores y adultos pueden presentar estreñimiento crónico y obstrucción intestinal. Otros problemas digestivos incluyen intususcepción, vólvulo, prolapso rectal y pancreatitis.
Otras Afecciones
Se observa infertilidad en aproximadamente el 98% de los hombres adultos. En mujeres, la fertilidad es algo menor. Otras complicaciones incluyen rinosinusitis crónica, osteopenia/osteoporosis, depresión, ansiedad, dolor crónico, apnea obstructiva del sueño, litiasis renal, nefropatía crónica, anemia ferropénica, hipoacusia neurosensorial y acúfenos.
Síntomas y Signos de la Fibrosis Quística
Aparato Respiratorio
Infecciones recurrentes o crónicas con tos, producción de esputo y sibilancias son frecuentes. La tos es el síntoma crónico más común. Con la progresión de la enfermedad, se observan retracciones intercostales, uso de músculos respiratorios accesorios, tórax en tonel, hipocratismo digital, cianosis y disminución de la tolerancia al ejercicio. El compromiso del tracto respiratorio superior puede incluir poliposis nasal y rinosinusitis crónica.
Las complicaciones pulmonares incluyen neumotórax, infección por micobacterias no tuberculosas, hemoptisis, aspergilosis broncopulmonar alérgica (ABPA) y cor pulmonale.
Aparato Digestivo
El íleo meconial puede ser el primer signo en aproximadamente el 20% de los recién nacidos afectados por FQ, manifestándose por distensión abdominal, vómitos y falta de eliminación de meconio. Algunos lactantes pueden desarrollar perforación intestinal.
En lactantes sin íleo meconial, el retraso en recuperar el peso de nacimiento y el aumento de peso inadecuado pueden anunciar el comienzo de la enfermedad. La insuficiencia pancreática se manifiesta clínicamente en etapas tempranas con deposiciones frecuentes de heces esteatorréicas, distensión abdominal y patrón de crecimiento deficiente.
El prolapso rectal puede ocurrir en lactantes y niños pequeños no tratados. Las manifestaciones clínicas secundarias a deficiencia de vitaminas liposolubles A, D, E y K también son comunes.
Tratamiento y Manejo de la Fibrosis Quística
El tratamiento de la FQ es multidisciplinario y de por vida, enfocado en:
- Problemas pulmonares: Antibióticos para prevenir y tratar infecciones, medicamentos inhalados para abrir vías respiratorias, terapias para diluir el moco (enzima DNAasa, solución salina hipertónica), vacunas antigripal y antineumocócica, y en algunos casos, trasplante de pulmón u oxigenoterapia.
- Problemas intestinales y nutricionales: Dieta especial rica en proteínas y calorías, enzimas pancreáticas con cada comida, y suplementos vitamínicos (A, D, E, K).
- Medidas generales: Evitar humo, polvo, químicos, tomar suficientes líquidos y hacer ejercicio regularmente.
La calidad de vida de los niños con FQ depende de las complicaciones al momento del diagnóstico y la accesibilidad a tratamientos a menudo costosos. El cribado neonatal es fundamental para mejorar el pronóstico.
Estudios y Resultados en el Cribado Neonatal de FQ
Los estudios retrospectivos analíticos de las concentraciones de tripsina inmunorreactiva (TIR) en RN con cribado neonatal positivo para FQ han revelado diferencias significativas. Los RN con FQ presentan cifras de TIR significativamente más elevadas que los sanos, portadores sanos o aquellos con diagnóstico inconcluso (CFSPID). La prematuridad y la hospitalización también pueden influir en los niveles de TIR.
Existe una correlación directa entre los niveles de TIR y el test del sudor en pacientes afectos de FQ y CFSPID. La curva ROC permite calcular un valor de corte de TIR para el diagnóstico de FQ, con alta sensibilidad y especificidad moderada.
La reciente introducción del cribado neonatal de FQ mediante la determinación de TIR ha mejorado la calidad de vida y supervivencia de los pacientes, permitiendo un diagnóstico precoz. Sin embargo, la baja especificidad de esta prueba se traduce en un elevado número de falsos positivos, lo que genera ansiedad en las familias y costes adicionales por estudios genéticos y test del sudor.
tags: #fibrosis #quistica #pruebas #metabolicas