La insulina y el glucagón, ambas hormonas esenciales liberadas por los islotes pancreáticos, desempeñan un papel crucial en la regulación de la homeostasis y el metabolismo de los carbohidratos. Un delicado equilibrio entre la secreción de estas dos hormonas mantiene los niveles de glucosa en plasma dentro de un rango fisiológico estrecho. Investigaciones clínicas han indicado que un aumento inapropiado del glucagón, conocido como hiperglucagonemia, contribuye significativamente a la patogénesis de la diabetes. Sin embargo, durante décadas, la investigación se ha centrado predominantemente en el papel de la insulina y en las terapias de reemplazo de esta hormona. Este artículo resume los avances recientes en la comprensión de las acciones del glucagón, su implicación en la fisiopatología de la diabetes y el potencial terapéutico de la inhibición de su receptor para el tratamiento de esta enfermedad.
Introducción a los Islotes Pancreáticos y sus Hormonas
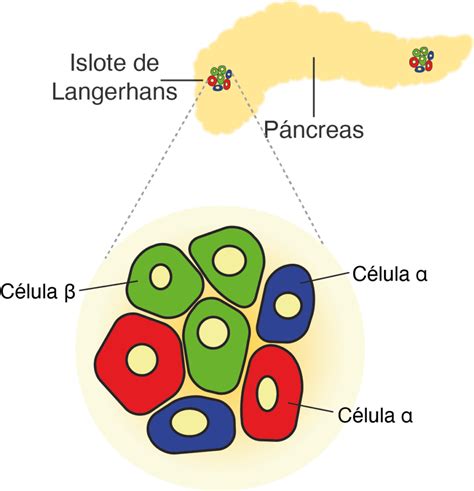
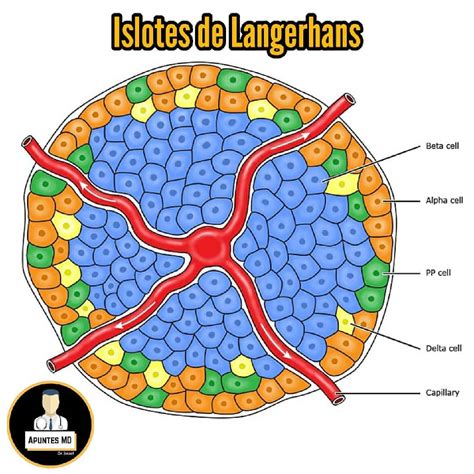
La porción endocrina del páncreas está compuesta por los islotes de Langerhans, estructuras distribuidas por todo el órgano, que representan aproximadamente el 2% de su volumen total. Dentro de estos islotes, las células α y β son las principales responsables de la síntesis y secreción de glucagón e insulina, respectivamente. Estas dos hormonas son los reguladores primarios de la homeostasis y el metabolismo de la glucosa.
Desde el descubrimiento de la insulina por Banting y Best en 1921, se le ha otorgado un papel central y casi exclusivo en la fisiopatología de la diabetes mellitus. No obstante, la evidencia sugiere que la disfunción en las interacciones paracrinas dentro del islote podría ser clave en el desarrollo de trastornos del metabolismo de los carbohidratos y formar parte integral de los factores que desencadenan la diabetes mellitus.
Este artículo de revisión se centrará en las acciones biológicas del glucagón, su contribución a la fisiopatología de la diabetes mellitus y las posibles intervenciones farmacológicas para modular sus efectos en el tratamiento de esta patología.
Síntesis y Secreción de Glucagón
El glucagón es un polipéptido de 29 aminoácidos secretado por las células α del islote pancreático. Su origen es el preproglucagón, un precursor de 180 aminoácidos que, tras su procesamiento, da lugar a glucagón, péptido similar al glucagón (GLP) tipo 1 y 2, oxintomodulina y glicentina. El procesamiento diferencial del preproglucagón en distintos tejidos, mediado por las prohormonas convertasas (PC1 y PC2), determina la producción predominante de glucagón en las células α pancreáticas.
La secreción de glucagón, al igual que la de insulina, está fundamentalmente regulada por los niveles de glucosa en plasma. Una disminución de la glucemia estimula el canal de potasio dependiente de ATP (KATP), lo que despolariza la membrana celular y permite la apertura de canales de sodio (Na+) y calcio (Ca+2). El aumento del calcio intracelular promueve la exocitosis de los gránulos de glucagón. Por el contrario, un aumento de la glucemia incrementa el ATP intracelular, cerrando los canales KATP, cesando la despolarización y, por ende, la secreción de glucagón.
La insulina ejerce un efecto supresor sobre la secreción de glucagón en las células α. Actúa hiperpolarizando la membrana a través del canal KATP y promoviendo la translocación de un receptor de GABA a la membrana celular, que, al interactuar con el GABA secretado por las células β, también induce hiperpolarización y suprime la secreción de glucagón. Estudios en ratones con deleción selectiva del receptor de insulina en células α (αIRKO) han demostrado hiperglucemia y hiperglucagonemia posprandial, subrayando el papel modulador de la insulina en la secreción de glucagón.
El zinc también se ha asociado con la modulación de la secreción de glucagón, pudiendo inducir hiperpolarización de la membrana de forma sinérgica con la insulina. Otros factores que regulan la secreción de glucagón incluyen el GLP-1, GLP-2, ácidos grasos, el sistema nervioso autónomo y los aminoácidos circulantes.
Mecanismo de Acción y Efectos Biológicos del Glucagón
El glucagón ejerce sus efectos al unirse a su receptor, un receptor acoplado a proteína G con siete dominios transmembrana, expresado principalmente en el hígado y riñón, y en menor medida en el corazón, adipocitos, páncreas endocrino, cerebro y tracto gastrointestinal. En el páncreas endocrino, el receptor de glucagón se expresa en las células β, sugiriendo una interacción paracrina bidireccional entre las células α y β. En ratones transgénicos con sobreexpresión del receptor de glucagón en células β, se observó un aumento en la secreción de insulina dependiente de glucosa y una mejoría en la tolerancia a los carbohidratos.
La unión del glucagón a su receptor activa la adenilciclasa, incrementando el AMPc intracelular y, consecuentemente, la proteína quinasa A (PKA). La PKA fosforila enzimas clave que desencadenan las acciones biológicas del glucagón. Adicionalmente, el glucagón se ha implicado en vías de señalización como la de la proteína quinasa asociada a mitógenos (MAPK) y la proteína quinasa activada por AMP (AMPK).
Efectos Hepáticos del Glucagón
A nivel hepático, el glucagón promueve la liberación de glucosa mediante la inhibición de la síntesis de glucógeno y la estimulación de la glucogenólisis y la gluconeogénesis. Favorece la captación de aminoácidos (alanina, glicina, prolina) que sirven como sustrato para la gluconeogénesis.
Efectos en el Adipocito
En el adipocito, el glucagón, a través de la lipasa sensible a hormonas, media la degradación de triglicéridos en ácidos grasos libres y glicerol. Aunque no modifica los niveles transcripcionales de esta enzima, aumenta la liberación de glicerol, que puede servir como sustrato para la gluconeogénesis. Asimismo, el glucagón inhibe la lipogénesis al reducir las concentraciones de malonil-CoA, lo que favorece la cetosis al activar la carnitina-palmitoil-transferasa, permitiendo la entrada de ácidos grasos a las mitocondrias para su oxidación a cuerpos cetónicos.
Citoarquitectura del Islote de Langerhans en la Diabetes Mellitus
En ratones, las células β forman el núcleo del islote, mientras que las células α se ubican en la periferia. La microcirculación en el islote permite que la insulina de las células β influya en la secreción de glucagón por las células α. En islotes humanos, la distribución celular es menos definida, pero se observa que aproximadamente el 71% de las células β están en contacto con células no β, facilitando las interacciones paracrinas.
Las células β pancreáticas, estimuladas por la hiperglucemia, responden con una secreción bifásica de insulina. La primera fase, rápida y de corta duración, aunque representa solo el 7% de la insulina liberada, se acompaña de una disminución recíproca de la glucagonemia, sugiriendo que esta rápida secreción de insulina actúa como una señal paracrina para inhibir la secreción de glucagón.
Diabetes Mellitus Tipo 1
En la diabetes mellitus tipo 1 (DM1), la destrucción autoinmune de las células β priva a las células α de la inhibición por insulina, resultando en hiperglucagonemia. Esta condición tiene efectos catabólicos severos y puede conducir a cetoacidosis diabética. Es notable que la concentración de insulina necesaria para suprimir la secreción de glucagón en las células α es 50 veces mayor que la requerida para la homeostasis glucémica en tejidos periféricos. Esto plantea la hipótesis de que la insulinización en DM1 podría no ser suficiente para suprimir completamente las células α sin causar "sobreinsulinización" en tejidos periféricos, con posibles efectos deletéreos.
Diabetes Mellitus Tipo 2
En las etapas iniciales de la diabetes mellitus tipo 2 (DM2), la citoarquitectura del islote es similar a la de pacientes no diabéticos. Sin embargo, la DM2 se caracteriza por la disfunción de las células β debido a la glucolipotoxicidad, que eventualmente conduce a apoptosis y pérdida de masa celular β. Esto deja a las células α sin la inhibición paracrina de la insulina. Dado que la fase rápida de secreción insulínica, crucial para suprimir las células α, se afecta precozmente en la DM2, la restitución de esta fase podría mejorar el control metabólico al inhibir la hiperglucagonemia relativa observada en estos pacientes.
El Papel del Glucagón en la Etiopatogenia de la Diabetes Mellitus
Aunque la diabetes mellitus se considera un "continuum", la relevancia del glucagón en su patogénesis varía a lo largo de la enfermedad. Algunas investigaciones sugieren que la hiperglucagonemia no es un fenómeno precoz, mientras que otras la vinculan tempranamente con la diabetes, incluso con estados de resistencia a la insulina previos.
La disfunción de la célula β es un defecto clave en la patogénesis de la DM2. La resistencia de las células β a la insulina podría reflejarse en las células α, llevando a una incapacidad para controlar la secreción de glucagón mediada por insulina. Esto apoya la teoría "bihormonal pancreática" de la diabetes propuesta por Unger y Orci, sugiriendo que una célula α "irrefrenable" podría ser un defecto temprano o concomitante a la disfunción secretora de insulina.
Estudios recientes en ratones knockout para el receptor de glucagón han demostrado que, tras la destrucción de las células β, se mantiene una tolerancia glucídica normal y se suprime la cetogénesis a pesar de la ausencia de insulina. Esto sugiere que el hígado, si no está expuesto a la acción del glucagón, podría tener un papel marginal en la regulación glucémica en ausencia de insulina. La especulación sobre la producción de glucagón intestinal en pacientes pancreatectomizados podría explicar la severidad del fenotipo diabético observado en esta población.
Insulina: Estructura, Síntesis y Mecanismos de Acción
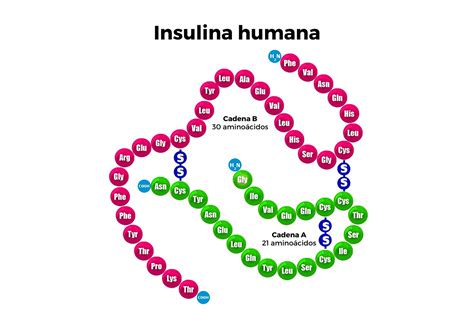
La insulina es una hormona polipeptídica compuesta por dos cadenas: la cadena A (21 aminoácidos) y la cadena B (30 aminoácidos), unidas por dos puentes disulfuro. La forma activa de la insulina es monomérica, circulante y capaz de unirse al receptor. En los gránulos secretorios de las células β, la insulina se almacena en forma de hexámeros coordinados con zinc.
La síntesis de insulina comienza con la transcripción del gen de la insulina, ubicado en el cromosoma 11p15.5, que da lugar a preproinsulina. Esta se procesa en el retículo endoplasmático a proinsulina, y posteriormente en el aparato de Golgi, se elimina el péptido C para formar insulina madura, que se almacena en gránulos secretorios.
La secreción de insulina puede depender de los canales KATP, un mecanismo directamente relacionado con la glucemia. El aumento de la relación ATP/ADP, debido a la utilización de glucosa, cierra los canales KATP, despolariza la membrana y permite la entrada de Ca+2, desencadenando la secreción de insulina. Alternativamente, las incretinas (GIP y GLP-1) actúan a través de receptores acoplados a proteínas G, aumentando el AMPc y activando la proteína quinasa A y Epac 2, lo que también libera insulina.
La depuración de insulina ocurre en células sensibles a la hormona, principalmente en hígado, riñón y músculo esquelético. La insulina es la hormona hipoglucemiante principal, promoviendo el transporte de glucosa a través del transportador GLUT4 en tejido adiposo, músculo y corazón. A diferencia de otros transportadores de glucosa (como GLUT3 en el cerebro), GLUT4 es sensible a la insulina.
Efectos Metabólicos de la Insulina
La insulina es fundamental para la síntesis neta de glucógeno hepático, aunque el transporte de glucosa al hepatocito no está regulado por insulina. Favorece la expresión de genes de lipogénesis de novo en hígado y tejido adiposo, y promueve la captación de triglicéridos circulantes. En el adipocito blanco, la insulina suprime potentemente la lipólisis, principalmente a través de la fosfodiesterasa 3B (PDE3B).
La insulina también ejerce otras funciones pro-lipogénicas, activando SREBP-1c y su programa transcripcional lipogénico en hepatocitos y adipocitos. Estos efectos sobre la expresión génica pueden ser duraderos, tardando semanas o meses en normalizarse.
Una dieta rica en carbohidratos, al aumentar la glucemia, promueve la secreción de insulina, lo que activa la síntesis y acumulación de grasa. En ausencia de secreción de insulina (glucemia basal), la síntesis de grasa no se activa y la piruvato carboxilasa puede desempeñar su papel anaplerótico para la degradación de grasas. La función de la insulina está intrínsecamente ligada al mantenimiento de niveles bajos de glucosa en sangre, más que a las necesidades energéticas celulares.
Tratamiento con Insulina y Análogos
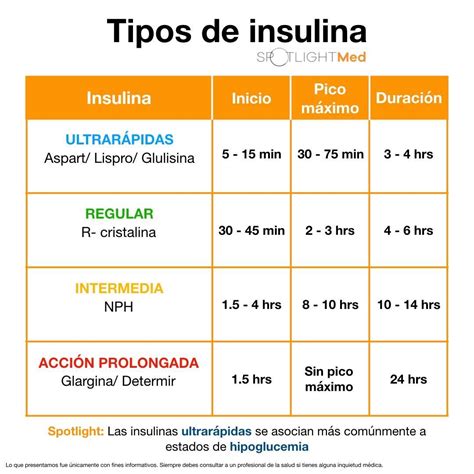
La insulina es el tratamiento más eficaz para reducir la hiperglucemia, siendo imprescindible en DM1 y a menudo necesaria en DM2. El tratamiento con insulina busca reproducir la secreción fisiológica, con picos posprandiales y niveles basales estables. Sin embargo, la administración subcutánea difiere de la administración fisiológica a la vena porta, que irriga directamente el hígado.
El estudio DCCT demostró que el tratamiento intensivo con insulina en DM1 reduce las complicaciones microvasculares, pero aumenta el riesgo de hipoglucemias. Para mejorar el perfil farmacocinético y farmacodinámico, se han desarrollado análogos de insulina de acción rápida (AIAR) y prolongada (AIAP).
Los AIAR de primera generación (lispro, aspart, glulisina) y la nueva generación como faster aspart, presentan modificaciones moleculares que aceleran su disociación monomérica y absorción. Los AIAP de primera (glargina U100, detemir) y segunda generación (degludec, glargina U300) ofrecen perfiles de liberación más predecibles y de mayor duración, reduciendo el riesgo de hipoglucemias nocturnas.
La estrategia basal-bolus, utilizando múltiples dosis de insulina (MDI) o infusión subcutánea continua de insulina (ISCI), es la que mejor reproduce la secreción fisiológica, aunque requiere múltiples inyecciones diarias. La elección de la insulina prandial y basal depende de las necesidades individuales del paciente y del control glucémico deseado.
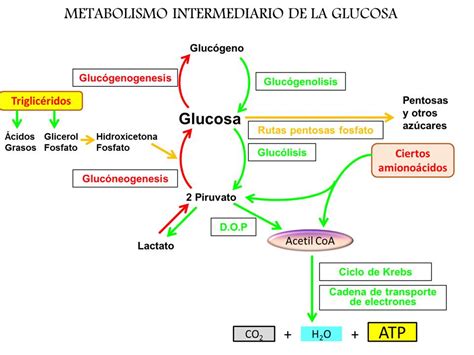
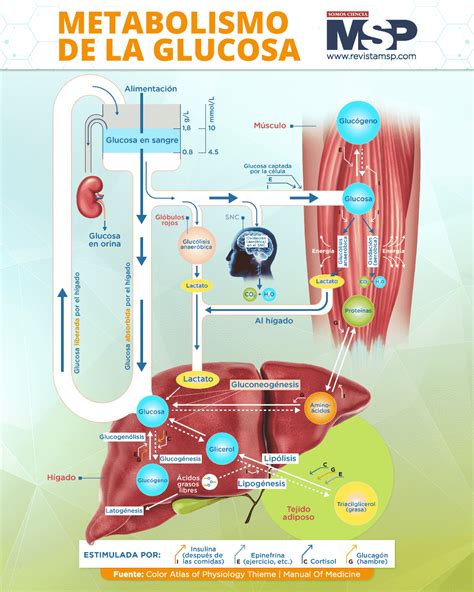
Metabolismo del Glucógeno
El glucógeno es un polisacárido animal que sirve como reserva energética, almacenado principalmente en el hígado y los músculos. Su metabolismo implica dos procesos principales: la glucogenólisis (degradación) y la glucogenogénesis (síntesis).
Las reservas de glucógeno muscular se agotan rápidamente durante el ejercicio intenso, mientras que las hepáticas pueden durar entre 12 y 24 horas durante el ayuno. El glucógeno hepático (aproximadamente el 10% del peso del hígado) es crucial para mantener la glucosa en sangre, gracias a la enzima glucosa-6-fosfatasa que permite la liberación de glucosa a la circulación.
La glucogenólisis inicia con la acción de la glucógeno fosforilasa, que retira unidades de glucosa en forma de glucosa-1-fosfato. La enzima desramificante actúa sobre las ramificaciones del glucógeno. La síntesis de glucógeno (glucogenogénesis) implica la formación de UDP-glucosa como sustrato activo.
El metabolismo del glucógeno está controlado por tres hormonas principales: el glucagón (células α pancreáticas), que actúa en el hígado; la adrenalina (médula adrenal), que actúa en músculo e hígado; y la insulina, que promueve la síntesis de glucógeno.
Glucagón y Complicaciones Renales y Cardiovasculares
Las complicaciones renales y cardiovasculares son manifestaciones clínicas significativas de la diabetes mellitus. La infusión sistémica de glucagón ha demostrado inducir hiperfiltración glomerular, un proceso que podría estar mediado por su receptor en una vía dependiente de AMPc.
tags: #insulina #metabolismo #glucogeno