Los fármacos anticonvulsivantes son ampliamente utilizados en pediatría para el tratamiento de crisis convulsivas. Estos medicamentos actúan sobre el sistema nervioso central a diferentes niveles, como canales iónicos, unión sináptica, mediadores sinápticos y conducción del impulso, con el objetivo de disminuir las descargas eléctricas responsables de las convulsiones. Sin embargo, su acción sobre el sistema nervioso central se acompaña de una serie de efectos metabólicos diversos, algunos de los cuales se correlacionan con manifestaciones clínicas específicas.
Los anticonvulsivantes se clasifican en dos grupos: antiguos y nuevos. Los fármacos anticonvulsivantes antiguos más comunes incluyen el fenobarbital, el ácido valproico, la carbamazepina, la fenitoína y la primidona. De estos, el fenobarbital, el ácido valproico y la carbamazepina son los más utilizados. La fenitoína y la primidona se reservan para situaciones de urgencia en Unidades de Cuidados Intensivos (UCI).
Alteraciones en el metabolismo óseo-mineral
Los fármacos anticonvulsivantes pueden provocar alteraciones significativas en el metabolismo óseo-mineral. Los anticonvulsivantes de acción prolongada presentan un mayor riesgo de desmineralización ósea y fracturas en huesos predispuestos. Este riesgo se ve exacerbado por factores como el déficit de vitamina D, la hipocalcemia y el hiperparatiroidismo secundario.
Se ha propuesto un mecanismo para el desarrollo de osteopenias mediado por el receptor Pregnano X (PXR), el cual está relacionado con el metabolismo de la vitamina D. La vitamina D, una hormona precursora de la dieta, se hidroxila primero en el hígado para formar 25-Hidroxicolecalciferol (25(OH)D3) y luego en el riñón para producir la forma activa, 1,25-dihidroxivitamina D3 (1,25(OH)2D3), que promueve la absorción de calcio en el intestino. Una vez activa, se une al receptor de Vitamina D (VDR), formando un complejo con el receptor X del ácido retinoico (RXR) para ejercer sus funciones. La inactivación de estas hormonas ocurre a través del receptor PXR, que puede ser activado por ciertos fármacos.
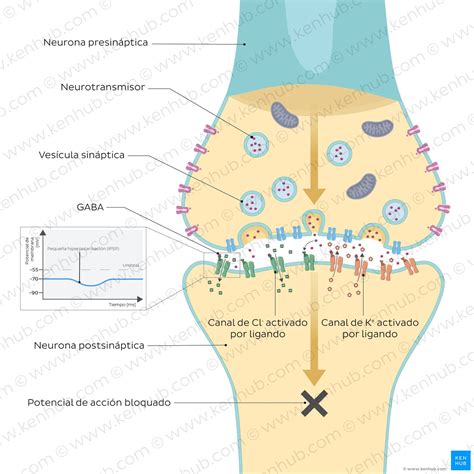
La hipótesis actual sugiere que fármacos como el fenobarbital, la fenitoína y la carbamazepina interfieren con la proteína VDR, impidiendo la inactivación de la hormona. Esto favorece la acción del receptor PXR, lo que lleva a la inactivación de la vitamina D y a una menor absorción de calcio intestinal. Aunque se ha sugerido la suplementación con vitamina D para mejorar estas condiciones, la evidencia científica, según una revisión de la base de datos Cochrane, no es suficiente para respaldar esta medida de forma concluyente. Se necesitan estudios controlados y aleatorizados para evaluar su utilidad real en pacientes que utilizan anticonvulsivantes.
Estudios previos en adultos (Christiansen, 1973) mostraron un contenido mineral óseo significativamente superior en pacientes epilépticos que recibían suplementos de vitamina D. Un estudio posterior en niños (Christiansen, 1975) también sugirió un efecto beneficioso, aunque con limitaciones metodológicas. La falta de confirmación posterior y la ausencia de datos sobre dosis efectivas hacen que el uso de vitamina D en estos casos sea, en gran medida, empírico.
Anticonvulsivantes y homocisteína
Ciertos anticonvulsivantes pueden afectar el metabolismo de la homocisteína, un aminoácido relacionado con el riesgo de accidentes vasculares. La homocisteína es normalmente metabolizada a metionina por la enzima metionina sintetasa, con la vitamina B12 como cofactor, en un proceso que involucra las vitaminas B6 y ácido fólico y es crucial para la síntesis de ADN y la metilación.
La carbamazepina y el ácido valproico interfieren con la conversión de homocisteína a metionina, elevando sus niveles en sangre. Estos niveles elevados de homocisteína tienen efectos aterogénicos y epileptogénicos. Se postula que esta interferencia ocurre a través de la alteración del metabolismo del ácido fólico y la vitamina B12.
Efectos de anticonvulsivantes específicos en el metabolismo
Carbamazepina
La carbamazepina puede causar alteraciones hepáticas, manifestadas por pruebas funcionales anormales, colestasis, ictericia hepatocelular, hepatitis e, infrecuentemente, falla hepática. También se han observado alteraciones del metabolismo lipídico, aunque los efectos sobre los lípidos sanguíneos y la función hepática son variables y no consistentes entre estudios. Se ha descrito un aumento del colesterol total y del LDL, posiblemente debido a la inducción de enzimas microsomales hepáticas que alteran el metabolismo lipídico. Se cree que el fenobarbital y el ácido valproico podrían provocar efectos similares. Adicionalmente, la carbamazepina puede causar pancreatitis.
Ácido valproico
El ácido valproico, o ácido 2-propil-pentanoico, es un ácido graso ramificado que puede influir en varios parámetros bioquímicos. Produce un aumento biológico de amoniaco, bilirrubina, colesterol HDL, testosterona y su proteína transportadora (globulina de unión a hormonas sexuales). Por otro lado, disminuye biológicamente la albúmina, la testosterona libre, los cuerpos cetónicos, el colesterol LDL, los triglicéridos y las hormonas prolactina, tirosina y triyodotironina (T3).
Aproximadamente el 80% del ácido valproico se metaboliza en el hígado por glucuronización. El 20% restante se elimina por excreción urinaria o mediante la beta oxidación mitocondrial, la misma vía que utilizan los ácidos grasos. Al utilizar la vía de entrada a la mitocondria dependiente de la L-carnitina, el ácido valproico puede capturar L-carnitina, bloqueando su propia metabolización y forzando una vía alternativa: la sigma oxidación en el citoplasma. Este proceso produce ácido 2-propil-4-pentanoico, una molécula asociada con la toxicidad del ácido valproico.
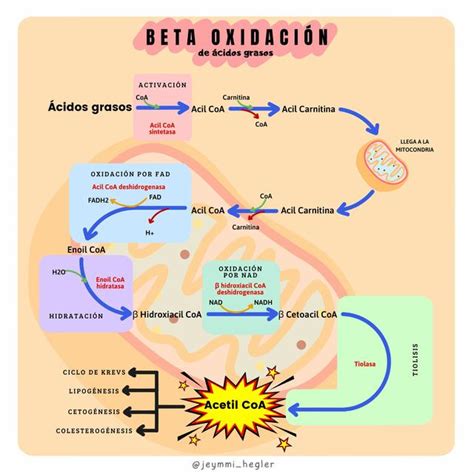
La beta oxidación implica la unión del ácido valproico a la coenzima A para entrar en las mitocondrias, seguido de un intercambio por carnitina para acceder a la matriz mitocondrial. La captura de carnitina por el ácido valproico puede afectar la generación de energía celular, especialmente durante la metabolización hepática de las grasas, e impedir la salida de carnitina de la mitocondria.
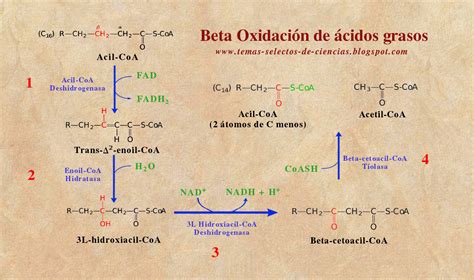
El ácido valproico que no se metaboliza por beta oxidación sigue la vía de sigma oxidación. El producto de esta vía, el ácido-2-propil-4-pentanoico, inhibe la enzima carbamoil fosfato sintetasa I (CPS I), esencial para el ciclo de la urea en el hígado. La inhibición de CPS I puede llevar a la acumulación de amonio, similar a lo que ocurre en la enfermedad del ciclo de la urea.
Los metabolitos del ácido valproico, al combinarse con acetil coenzima A, impiden la reposición de los depósitos de carnitina. La depleción de carnitina resultante puede causar:
- Reducción del transporte de ácidos grasos de cadena larga y de su beta oxidación.
- Desvío del metabolismo hacia la sigma oxidación, con producción del metabolito tóxico.
- Acumulación intracelular de amonio.
Otro efecto secundario del ácido valproico es el aumento de peso, para el cual se proponen varios mecanismos: aumento del apetito, acción directa sobre el hipotálamo, disminución de la termogénesis facultativa, y desarrollo de hiperinsulinemia y resistencia a la insulina. Estudios han observado en pacientes que usan ácido valproico un aumento de insulina y leptina, y una disminución de grelina y adiponectina, lo que favorecería la obesidad.
Fenitoína
La exposición prenatal a la fenitoína se asocia con un mayor riesgo de malformaciones congénitas, alteraciones del desarrollo, anomalías menores y mayores, problemas de crecimiento y retraso mental. En adultos, el uso de fenitoína ha demostrado fallos en la respuesta de insulina.
Frente a una infusión de glucosa, los individuos normales presentan una respuesta bifásica de insulina (rápida y lenta) para mantener la glucemia en rangos normales. Los pacientes que usan fenitoína muestran una respuesta de primera fase muy reducida y ausencia de respuesta de segunda fase, lo que los predispone a la hiperglucemia. Esto es particularmente problemático para pacientes con diabetes o resistencia a la insulina.
Topiramato
El topiramato, uno de los anticonvulsivantes más utilizados, ejerce su acción mediante varios mecanismos: bloqueo de canales de sodio dependientes de voltaje, modulación negativa de receptores de glutamato, aumento de la actividad de receptores GABAA e inhibición de la anhidrasa carbónica (isoenzimas II y IV). Esta última acción tiene efectos sobre el estado de los electrolitos.
El uso de topiramato se asocia con oligohidrosis (disminución de la producción de sudor) y hipertermia, lo que compromete el mecanismo normal de protección contra el sobrecalentamiento. En pacientes pediátricos, esto podría deberse al bloqueo de la anhidrasa carbónica, alterando la composición del sudor y el resultado de las pruebas de sudoración en pacientes con fibrosis quística.
La inhibición de la anhidrasa carbónica también produce acidosis metabólica, hipercloremia sin anion gap y efectos a nivel del túbulo renal. El uso prolongado de topiramato puede alterar el crecimiento y aumentar el riesgo de litiasis renal, por lo que se debe evitar su administración en niños pequeños.
Lamotrigina
La lamotrigina está autorizada como monoterapia en mayores de 12 años y en terapia combinada desde los dos años. Actúa sobre el metabolismo del ácido fólico al inhibir la dihidrofolato reductasa. Se recomienda la suplementación con ácido fólico para reducir el riesgo de trastornos relacionados y evitar el uso concomitante de otros inhibidores del ácido fólico, como el cotrimoxazol.
Levetiracetam
La FDA no autoriza el uso de levetiracetam en menores de 16 años. Su mecanismo de acción implica la unión a una proteína de la vesícula sináptica, la SV2A.
Interacción entre anticonvulsivantes y anticonceptivos orales
Varios anticonvulsivantes, incluyendo fenobarbital, carbamazepina, fenitoína, primidona, oxicarbazepina, lamotrigina, topiramato y felbamato, pueden interferir con la efectividad de los anticonceptivos orales, disminuyendo su eficacia.
Estudios sobre lípidos y anticonvulsivantes
La terapia con ácido valproico se asocia con riesgo de déficit de carnitina. Para investigar el impacto del uso de FAEIE (Fármacos Antiepilépticos que Inducen Enzimas Hepáticas) en los niveles de lípidos séricos en pacientes ancianos y comparar con adultos más jóvenes, se realizó un estudio. Se incluyeron 235 pacientes adultos con epilepsia tratados con FAEIE y 213 sin ellos, divididos por edad. Los pacientes que tomaban carbamazepina y fenitoína se clasificaron como grupo FAEIE.
Algunos estudios han indicado que los medicamentos anticonvulsivantes, especialmente los inductores enzimáticos, pueden elevar las concentraciones de lipoproteínas de alta densidad (HDL), lo cual podría ser un efecto colateral beneficioso que disminuya el riesgo cardiovascular. Dada la cronicidad del tratamiento antiepiléptico, se ha investigado la influencia de diferentes tratamientos sobre el perfil lipídico plasmático.
En un estudio con 385 individuos (75 sanos como control y 310 epilépticos en tratamiento crónico), se midieron niveles de colesterol total, HDL, triglicéridos y apoproteínas A y B. Todos los tratamientos valorados modificaron los lípidos y apoproteínas. Los efectos más favorables se observaron en mujeres y en sujetos menores de 15 años.
La carbamazepina y el ácido valproico mostraron los efectos más positivos: la carbamazepina elevó el HDL y el ácido valproico disminuyó el LDL, ambos reduciendo el cociente CT/HDL-C (índice de riesgo de arteriosclerosis). Las cuatro terapias estudiadas aumentaron significativamente la apoproteína A, especialmente la carbamazepina. El ácido valproico fue el único que indujo modificaciones relevantes en la apoproteína B, produciendo un descenso.
Fármacos antiepilépticos como la carbamazepina, fenitoína, fenobarbital, primidona, topiramato, eslicarbazepina, oxcarbazepina y rufinamida, clasificados como inductores enzimáticos, se asocian con un incremento significativo de eventos isquémicos cardíacos y cerebrales, incluso tras corta exposición. Estos fármacos también aumentan el colesterol, la proteína C reactiva y otros marcadores de riesgo cardiovascular, además de interactuar con otros medicamentos.
Antiepilépticos
Monitorización Terapéutica de Fármacos Antiepilépticos
La monitorización terapéutica de niveles séricos (TDM) de fármacos antiepilépticos consiste en la determinación periódica de sus concentraciones en suero para optimizar la eficacia y seguridad del tratamiento. Este procedimiento, que comenzó hace unos 30 años, ha mejorado la utilización racional de estos fármacos.
La TDM se fundamenta en varias razones:
- La valoración clínica de los efectos farmacológicos es a menudo difícil y lenta.
- Existe una mejor correlación entre el efecto farmacológico y el nivel sérico del antiepiléptico que con la dosis prescrita, debido a diferencias farmacocinéticas interindividuales y a la cinética no lineal de algunos compuestos.
- Muchos anticonvulsivantes presentan un margen terapéutico estrecho, donde las concentraciones mínima eficaz y mínima tóxica están próximas.
- La valoración clínica de la toxicidad puede ser complicada en neonatos, pacientes con alteraciones neurológicas o retraso psicomotor.
- Es necesario prevenir reacciones tóxicas graves.
- Para varios antiepilépticos, su efecto es proporcional a la concentración en el sistema nervioso central.
Los antiepilépticos tradicionales tienen un intervalo óptimo de concentraciones séricas donde se logra eficacia sin toxicidad. Sin embargo, este intervalo no debe aplicarse rígidamente de forma individualizada. Las tendencias actuales sugieren una interpretación más flexible de estos rangos, considerando cualquier concentración que no alcance el límite superior como potencialmente terapéutica, o estableciendo intervalos concéntricos para definir un rango óptimo y unos límites de concentraciones extremas tolerables.
Aspectos clínicos de la farmacocinética
Los antiepilépticos se administran generalmente por vía oral y sufren procesos de absorción, distribución, metabolización y excreción. Cada fármaco posee parámetros farmacocinéticos que lo individualizan.
Absorción
La absorción gastrointestinal suele ser buena, con una fracción de absorción alta, excepto en la carbamazepina. La velocidad de absorción varía (rápida para el ácido valproico, lenta para fenitoína y carbamazepina). El tiempo para alcanzar la concentración sérica máxima (Tmáx) depende de las velocidades de absorción y eliminación.
Factores que pueden modificar la absorción oral incluyen:
- Presencia de alimentos: suele disminuirla (valproato), pero puede aumentar la disolución de fármacos poco hidrosolubles como fenitoína o carbamazepina.
- Nutrición enteral: puede disminuirla (fenitoína).
- Preparado farmacéutico: pueden existir diferencias de biodisponibilidad entre preparados comerciales (fenitoína, carbamazepina, fenobarbital) e incluso con formulaciones magistrales.
- Dosis utilizada: la biodisponibilidad de carbamazepina y fenitoína disminuye al aumentar la dosis.
Distribución
Una vez absorbidos, los antiepilépticos se distribuyen por el organismo, uniéndose a proteínas plasmáticas (principalmente albúmina). El volumen de distribución aparente refleja la cantidad de fármaco en los tejidos en relación a la concentración sérica. Factores como la edad, el embarazo y la unión a proteínas influyen en este volumen.
Solo el fármaco libre (no unido a proteínas) penetra en los tejidos. La fenitoína y el ácido valproico se unen en alta proporción a proteínas séricas. La unión a proteínas puede disminuir por factores fisiológicos (embarazo, neonatos, ancianos), patológicos (hipoalbuminemia, nefropatía) o farmacológicos, aumentando la fracción libre y, a veces, la toxicidad.
Eliminación
La principal vía de eliminación es la biotransformación hepática. La excreción urinaria sin biotransformar es importante para primidona y fenobarbital. La metabolización de carbamazepina y primidona produce metabolitos activos que influyen en la eficacia y toxicidad.
La vida media es el parámetro más usual para estimar la eliminación, representando el tiempo necesario para que la concentración sérica disminuya a la mitad.
Tipos de cinética
Cinética Lineal
La intensidad y velocidad de los procesos farmacocinéticos no varían con el tiempo ni la dosis. Existe una relación lineal entre la dosis administrada y el nivel sérico estable alcanzado.
Cinética No Lineal
Las constantes farmacocinéticas varían con el tiempo o la dosis. Puede ser dependiente de la dosis (creciente o decreciente) o dependiente del tiempo.
- Dependiente de la dosis: Un ejemplo es la biotransformación de la fenitoína, que se satura y disminuye desproporcionadamente, aumentando el riesgo de toxicidad.
- Dependiente del tiempo: La carbamazepina es un ejemplo, donde la eliminación aumenta progresivamente durante el primer mes por autoinducción enzimática, lo que resulta en una vida media más larga tras dosis única que en tratamiento crónico.
Utilidad de la relación Dosis-Nivel Sérico
La variabilidad farmacocinética interindividual dificulta predecir la concentración sérica con una dosis determinada. La fenitoína y el valproato son particularmente problemáticos debido a su cinética dosis-dependiente, interacciones y alto grado de unión a proteínas séricas.
Factores que determinan la relación Dosis-Nivel Sérico
- Factores genéticos: Variabilidad en la actividad de enzimas metabolizadoras.
- Edad: El metabolismo hepático evoluciona con la edad, requiriendo ajustes de dosis en neonatos, niños y ancianos.
- Menstruación: Puede disminuir los niveles séricos de fenitoína.
- Embarazo: Modifica la albúmina sérica, la capacidad metabólica hepática y el líquido corporal.
- Parto: La biotransformación de antiepilépticos se normaliza en semanas.
- Fiebre: Aumenta la biotransformación y disminuye los niveles séricos de algunos antiepilépticos.
- Enfermedad hepática: Puede disminuir la capacidad metabólica, la unión a proteínas y aumentar el volumen de distribución, requiriendo ajustes de dosis.
- Enfermedad renal: Puede afectar la excreción y la unión a proteínas.
tags: #antiepilepticos #afectan #al #metabolismo #lipidico