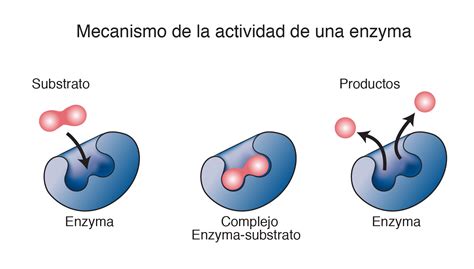
Los trastornos metabólicos hereditarios son un grupo de afecciones genéticas poco comunes en las que el cuerpo es incapaz de descomponer adecuadamente los alimentos en energía o de eliminar los productos de desecho. Estos trastornos se originan por mutaciones genéticas que afectan el funcionamiento de enzimas específicas, las cuales son cruciales para llevar a cabo miles de reacciones químicas dentro de las células. Cuando una enzima falta o no funciona correctamente, pueden ocurrir diversas alteraciones metabólicas, ya sea por la acumulación de sustancias intermedias tóxicas o por la deficiencia de productos esenciales.
Estos trastornos se transmiten de padres a hijos y, en la mayoría de los casos, siguen un patrón de herencia autosómica recesiva. Esto significa que una persona necesita heredar una copia del gen alterado de cada progenitor para desarrollar la afección. Si solo se hereda un gen alterado, la persona suele ser portadora pero no presenta síntomas.
Manifestaciones Clínicas de los Trastornos Metabólicos Hereditarios
Las manifestaciones clínicas de los trastornos metabólicos hereditarios pueden ser muy variadas y, a menudo, son inespecíficas, lo que puede dificultar su diagnóstico inicial, ya que a menudo se atribuyen a otras causas más comunes como infecciones.
Síntomas en el Período Neonatal
Los trastornos que se manifiestan en el período neonatal tienden a ser más graves. Las presentaciones clínicas comunes incluyen:
- Letargo
- Mala actitud alimentaria
- Vómitos
- Convulsiones
En esta etapa, los recién nacidos tienen un repertorio limitado de respuestas ante enfermedades graves. Los síntomas iniciales inespecíficos pueden incluir rechazo de la alimentación, vómitos explosivos, compromiso progresivo de la conciencia (desde letargia y somnolencia hasta coma profundo), convulsiones, compromiso hemodinámico e incluso muerte. También pueden presentarse edema cerebral y hemorragia intracraneal. Es importante destacar que los pacientes con errores innatos del metabolismo tienen un mayor riesgo de presentar infecciones, lo que puede enmascarar el diagnóstico.
Síntomas en Manifestaciones Tardías
Los trastornos que se manifiestan de forma más tardía suelen comprometer el crecimiento y el desarrollo. Sin embargo, también pueden presentarse vómitos, convulsiones y debilidad muscular. El retraso del crecimiento puede deberse a una disminución del anabolismo o un aumento del catabolismo, lo que puede resultar de una menor disponibilidad de sustratos generadores de energía o del uso ineficiente de energía o proteínas.
El retraso del desarrollo puede reflejar un déficit crónico de energía en el encéfalo, un aporte insuficiente de carbohidratos no energéticos o un déficit crónico de aminoácidos en el encéfalo.
Síntomas Neuromusculares
Los síntomas neuromusculares como convulsiones, debilidad muscular, hipotonía, mioclonías, dolor muscular, accidentes cerebrovasculares o coma pueden sugerir un déficit agudo de energía en el encéfalo o en el músculo. Asimismo, pueden reflejar la acumulación de compuestos tóxicos en el encéfalo o la degradación tisular (por ejemplo, rabdomiólisis y mioglobinuria).
Malformaciones Encefálicas Congénitas
Las malformaciones encefálicas congénitas pueden ser un reflejo de una menor disponibilidad de energía o de precursores críticos durante el desarrollo fetal.
Síntomas Neurovegetativos
Los síntomas neurovegetativos pueden deberse a hipoglucemia (causada por mayor consumo o menor producción de glucosa) o a acidosis metabólica. Algunos cuadros pueden presentar ambas condiciones simultáneamente.
Ictericia
La ictericia no fisiológica después del período neonatal suele indicar hepatopatía intrínseca, pero también puede ser manifestación de trastornos hereditarios del metabolismo.
Olores Inusuales
Los olores inusuales de los líquidos orgánicos son un signo característico de la acumulación de compuestos específicos. Por ejemplo:
- Olor a pies sudorosos: Acidemia isovalérica.
- Olor ahumado dulce: Enfermedad de la orina con olor a jarabe de arce.
- Olor rancio o a ratón: Fenilcetonuria.
- Olor a repollo hervido: Tirosinemia.
Cambio del Color de la Orina
En algunos trastornos, se observa un cambio del color de la orina al exponerla al aire, como marrón oscuro en la alcaptonuria o marrón violáceo en la porfiria.
Organomegalia
La organomegalia (agrandamiento de órganos) puede deberse a la ausencia de degradación de sustratos que se acumulan dentro de las células del órgano, como hepatomegalia en trastornos por depósito de glucógeno o cardiomegalia en la glucogenosis tipo II.
Alteraciones Oculares
Las alteraciones oculares pueden incluir cataratas o degeneración retiniana, y oftalmoplejía en ciertos defectos del metabolismo.
Diagnóstico de los Trastornos Metabólicos Hereditarios
El diagnóstico de los trastornos metabólicos hereditarios es un proceso multifacético que comienza con la sospecha clínica y se confirma mediante una serie de pruebas específicas.
Estudios Complementarios Iniciales
Cuando se sospecha un trastorno hereditario del metabolismo, la evaluación inicial incluye:
- Revisión de los resultados de las pruebas de cribado neonatales.
- Pruebas metabólicas básicas de tamizaje, que típicamente incluyen:
- Glucosa
- Electrolitos con cálculo de la brecha aniónica
- Hemograma completo y frotis periférico
- Pruebas hepáticas
- Concentraciones de amoníaco
- Niveles de aminoácidos séricos
- Análisis de orina
- Ácidos orgánicos en la orina
Análisis Específicos
La determinación de glucosa detecta hipoglucemia o hiperglucemia, y a veces requiere mediciones en momentos específicos respecto a las comidas.
La determinación de electrolitos ayuda a detectar acidosis metabólica y la brecha aniónica. En casos de acidosis metabólica, se puede corroborar mediante gases en sangre arterial. La acidosis sin hiato aniónico puede observarse en trastornos que causan daño tubular renal, mientras que la acidosis con hiato aniónico se presenta en trastornos con acumulación de ácidos titulables o acidosis láctica.
El hemograma completo y frotis periférico pueden detectar hemólisis o citopenia provocada por la acumulación de metabolitos.
Las pruebas de funcionalidad hepática identifican lesión o disfunción hepatocelular.
El aumento de las concentraciones de amoníaco es indicativo de defectos del ciclo de la urea, acidemias orgánicas y defectos de oxidación de ácidos grasos.
El análisis de orina detecta cetonuria; la ausencia de cetonas en presencia de hipoglucemia puede sugerir un defecto de oxidación de ácidos grasos o hiperinsulinismo.
Pruebas Específicas y de Confirmación
Cuando las pruebas de detección sistemática sugieren un trastorno metabólico, se indican pruebas más específicas, como:
- Medición de metabolitos de carbohidratos, mucopolisacáridos, aminoácidos y ácidos orgánicos mediante cromatografía y espectrometría de masa.
- Perfil de acilcarnitina en plasma y perfil de acilglicina en orina.
- Secuenciación de genes para identificar mutaciones conocidas.
- Biopsias (hepática, muscular) para análisis histopatológico y enzimático.
- Estudios de enzimas utilizando células sanguíneas o cutáneas.
Pruebas de Provocación
Aunque menos comunes debido a los avances en la detección de metabolitos, las pruebas de provocación se utilizan para detectar alteraciones bioquímicas mensurables en condiciones específicas. Estas incluyen pruebas de ayuno, pruebas de provocación con sustratos específicos (como fructosa o glucagón) y pruebas de esfuerzo.
¿Qué CRIBADOS se realizan en el recién nacido? ¿Para qué sirven?. CRIBADO NEONATAL
Tipos de Trastornos Metabólicos Hereditarios
Los trastornos metabólicos hereditarios se clasifican generalmente según el tipo de sustancia que el cuerpo tiene dificultades para procesar:
Trastornos del Metabolismo de los Aminoácidos
Afectan la forma en que el cuerpo descompone las proteínas. Ejemplos incluyen la fenilcetonuria (PKU), la enfermedad de la orina con olor a jarabe de arce y la homocistinuria. Estos trastornos a menudo requieren dietas especiales que limitan ciertas proteínas.
Trastornos del Metabolismo de los Carbohidratos
Incluyen la galactosemia (incapacidad para descomponer la galactosa) y la intolerancia hereditaria a la fructosa. Las enfermedades de almacenamiento de glucógeno también pertenecen a esta categoría, afectando el almacenamiento o la liberación de glucosa.
Trastornos del Metabolismo de las Grasas
Estas afecciones afectan la descomposición de las grasas para obtener energía. La deficiencia de acil-CoA deshidrogenasa de cadena media (MCAD) es un ejemplo común. Los trastornos de la oxidación de ácidos grasos de cadena larga pueden causar complicaciones graves.
Trastornos Mitocondriales
Las mitocondrias son las "centrales eléctricas" de las células. Cuando no funcionan correctamente, los órganos que requieren mucha energía (cerebro, corazón, músculos) pueden verse afectados. Estos trastornos son menos comunes pero pueden ser complejos.
Factores de Riesgo y Complicaciones
El principal factor de riesgo es tener padres que son portadores del mismo cambio genético. Antecedentes familiares, consanguinidad y ciertos orígenes étnicos pueden aumentar el riesgo. Las posibles complicaciones incluyen discapacidad intelectual, daño hepático o renal, problemas cardíacos, convulsiones, retrasos en el crecimiento y crisis metabólicas potencialmente mortales.
Prevención y Tratamiento
Si bien los trastornos metabólicos hereditarios no se pueden prevenir una vez que se nace con la afección, la detección neonatal permite un diagnóstico y tratamiento tempranos, lo que puede prevenir complicaciones graves. El asesoramiento genético es fundamental para parejas con factores de riesgo. El tratamiento se centra en ayudar al cuerpo a funcionar alrededor de la enzima deficiente, a menudo mediante dietas especiales, medicamentos, terapia de reemplazo enzimático o suplementos.
tags: #manifestaciones #trastorno #metabolico #hereditario