El metabolismo lipídico es de vital importancia para la vida, ya que los lípidos desempeñan roles esenciales en la estructura celular, la síntesis de hormonas y el almacenamiento de energía. El colesterol y los fosfolípidos son componentes fundamentales de todas las membranas celulares, garantizando su funcionalidad y supervivencia. Además, el colesterol es el precursor de las hormonas esteroideas. Por otro lado, los triglicéridos (TG) proporcionan los ácidos grasos, la principal fuente de energía para el organismo, y constituyen el componente principal del tejido adiposo, actuando como reserva energética crucial para mantener la actividad en periodos de ayuno y asegurar la supervivencia de la especie.
Existe una aparente paradoja en relación con el colesterol: si bien es indispensable para todas las células, incluyendo el cerebro, las células poseen la capacidad de sintetizarlo a partir de acetato si no lo reciben del exterior a través de las lipoproteínas de baja densidad (LDL). Sin embargo, el exceso de colesterol, derivado de alteraciones en el metabolismo lipídico, puede desencadenar la lesión aterosclerosa y, consecuentemente, la enfermedad arteriosclerosa, una de las principales causas de mortalidad a nivel mundial, especialmente en países desarrollados.
Dado su impacto en la patología, nos centraremos en el metabolismo del colesterol y los triglicéridos. Ambos lípidos son insolubles en agua (sangre), por lo que para su transporte se unen a proteínas específicas, las apolipoproteínas (apo), formando complejas estructuras denominadas lipoproteínas. Estas partículas presentan un núcleo compuesto por colesterol esterificado y TG, y una capa externa de colesterol libre, fosfolípidos y apolipoproteínas, lo que les confiere miscibilidad en medios acuosos.
Principales Lipoproteínas
Las principales lipoproteínas que circulan en el plasma son:
- Quilomicrones (QM): Partículas de gran tamaño (100-1200 nm) ricas en triglicéridos (más del 90%), transportan los lípidos de la dieta desde el intestino al resto del organismo. Su principal apolipoproteína es la Apo B-48.
- Remanentes de Quilomicrones (QMR): Partículas resultantes de la hidrólisis de los TG de los QM.
- Lipoproteínas de muy baja densidad (VLDL): Partículas de 45-100 nm, sintetizadas en el hígado, ricas en triglicéridos (aproximadamente 90%) y con Apo B-100 como proteína principal. Transportan los TG endógenos a los tejidos.
- Remanentes de VLDL (VLDLR) / Lipoproteínas de densidad intermedia (IDL): Partículas intermedias en el catabolismo de las VLDL, ricas en colesterol y TG.
- Lipoproteínas de baja densidad (LDL): Partículas de 20-25 nm, ricas en colesterol (60-70% del colesterol plasmático), con Apo B-100 como proteína principal. Transportan colesterol a las células periféricas.
- Lipoproteína (a) [Lp(a)]: Similar a la LDL pero con una apolipoproteína adicional, la apo(a).
- Lipoproteínas de alta densidad (HDL): Partículas de 25-10 nm, con los fosfolípidos como lípido principal y Apo A-I como proteína principal. Son producidas por el hígado y el intestino.
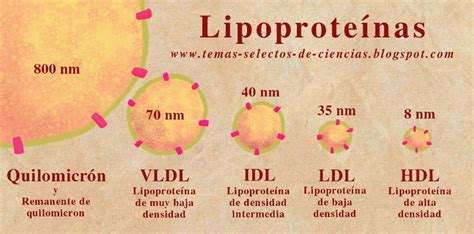
Metabolismo Exógeno de las Lipoproteínas
La vía exógena se refiere al transporte de lípidos desde el intestino (provenientes de la dieta y bilis) hacia el hígado y las células periféricas. Los TG alimentarios se hidrolizan en el intestino y, junto con el colesterol absorbido, se ensamblan en los enterocitos para formar los quilomicrones (QM), que se unen a la apoB48. Estos QM son secretados a la linfa intestinal y luego a la circulación general.
En la circulación, los QM interactúan con la lipoproteína lipasa (LPL), una enzima localizada en el endotelio vascular, que hidroliza los TG, liberando ácidos grasos libres para su captación por los tejidos (adiposo y muscular) como fuente de energía o para su almacenamiento.
Los QM, al perder TG, se modifican, transfiriendo colesterol y fosfolípidos a las HDL y adquiriendo apoE, transformándose en quilomicrones remanentes (QMR). Estas partículas son rápidamente captadas por el hígado a través de diversos receptores hepáticos.
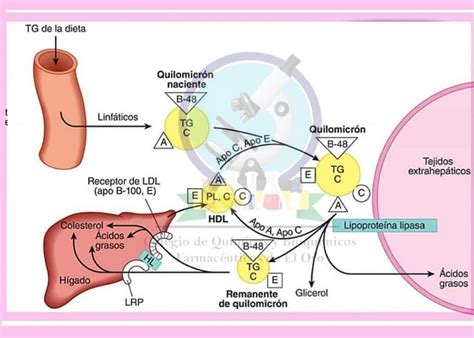
Metabolismo Endógeno de las Lipoproteínas
El hígado es central en el metabolismo lipídico, iniciando la vía endógena con la síntesis y secreción de las VLDL. Estas partículas, ricas en TG y con apoB100, se forman en el hígado mediante un proceso similar a la formación de QM, mediado por la enzima proteína de transporte microsómico (MTP).
Las VLDL circulantes son hidrolizadas por la LPL en los tejidos extrahepáticos, liberando ácidos grasos. Las partículas resultantes, empobrecidas en TG y enriquecidas en colesterol, se denominan VLDL remanentes (VLDLR) o IDL. Una porción de estas partículas es captada por el hígado, mientras que el resto se transforma en LDL por la acción de la lipasa hepática (HL) y la proteína transferidora de ésteres de colesterol (CETP).
Las LDL, ricas en colesterol, son captadas predominantemente por el hígado (aproximadamente 70%) a través del receptor de LDL (R-LDL), que reconoce la apoB100. El 30% restante es captado por las células periféricas. El número de R-LDL es saturable y su producción está regulada por factores de transcripción como los SREBP.
El receptor LDL participa en la endocitosis de las LDL. Tras la internalización, las LDL son dirigidas a los lisosomas para su degradación por la lipasa ácida lisosomal (LAL). El exceso de LDL no captada, junto con otras partículas lipídicas remanentes, puede atravesar la pared endotelial, iniciar el proceso aterosclerótico y acumularse en los macrófagos (receptor scavenger).
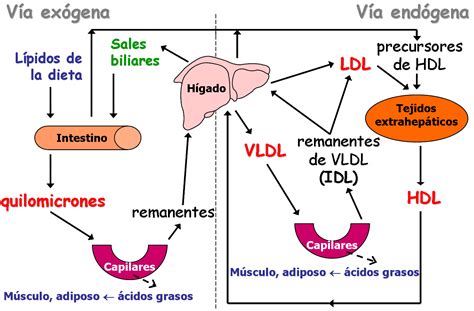
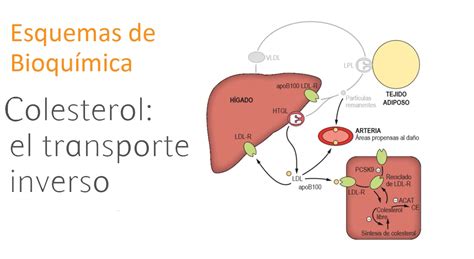
Transporte Inverso del Colesterol
El colesterol depositado en las células o en la pared arterial no puede ser metabolizado y eliminado directamente. La principal vía de eliminación es a través de las HDL, mediante el transporte inverso del colesterol. Las HDL nacientes, sintetizadas por el intestino y el hígado, captan colesterol no esterificado de los tejidos periféricos a través de transportadores como ABCA1 y SR-BI.
El colesterol captado es esterificado por la lecitina-colesterol aciltransferasa (LCAT), formando HDL maduras. Estas HDL enriquecidas en colesterol son captadas por los hepatocitos, principalmente a través del receptor SR-BI. El colesterol hepático se reutiliza para la síntesis de VLDL o se excreta por vía biliar como ácidos biliares o colesterol.
Tradicionalmente, las HDL se consideran el colesterol "bueno" por su papel protector cardiovascular. Sin embargo, las HDL son partículas complejas con múltiples funciones (antiinflamatoria, antioxidante, etc.) que pueden ser alteradas, pudiendo volverse perjudiciales.
Lisosomas y Metabolismo Lipídico
Los lisosomas son orgánulos celulares formados a partir del aparato de Golgi, que contienen una variedad de hidrolasas ácidas (lipasas, proteasas, nucleasas, etc.) encargadas de la degradación de material intracelular de origen externo (fagocitosis) o interno (autofagia). Su pH interno es ácido (aproximadamente 4.8), óptimo para la actividad de estas enzimas.
La membrana lisosómica está protegida de la acción de las enzimas por una capa de glucoproteínas y posee bombas de protones (v-ATPasa) que mantienen el pH ácido. También contienen proteínas de transporte que permiten la salida de los productos de la digestión al citosol.
Tipos de Lisosomas
- Lisosomas primarios: Vesículas recién formadas desde el Golgi, que contienen solo enzimas hidrolíticas y esperan fusionarse con material a degradar.
- Lisosomas secundarios: Resultado de la fusión de un lisosoma primario con una vesícula que contiene material a digerir (fagolisosomas o autofagolisosomas). Contienen tanto enzimas como sustratos.
- Cuerpos residuales: Material no digerible que queda tras la digestión en el lisosoma secundario. Pueden ser exocitados o acumularse en el citosol.
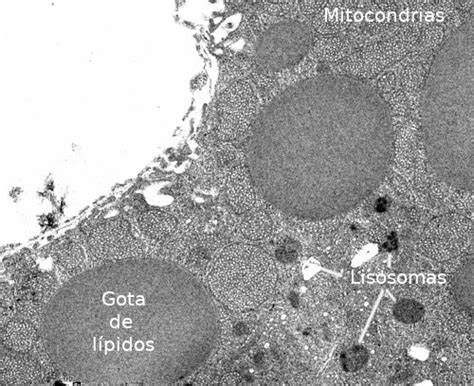
Rol de los Lisosomas en el Metabolismo Lipídico
Los lisosomas desempeñan un papel crucial en la degradación de lípidos, especialmente a través de la acción de la lipasa ácida lisosomal (LAL). La LAL es fundamental para la hidrólisis del colesterol esterificado y los triglicéridos acumulados en el interior de los lisosomas.
La deficiencia de LAL conduce a enfermedades como la enfermedad de Wolman (forma severa de inicio temprano) y la enfermedad de almacenamiento de ésteres de colesterol (CESD) (forma de inicio más tardío), caracterizadas por la acumulación progresiva de ésteres de colesterol y triglicéridos en diversos tejidos, como el hígado, bazo, ganglios linfáticos y paredes vasculares.
Estas enfermedades resultan en disfunción hepática, dislipidemia severa, xantomas, aterosclerosis prematura y, en casos severos, alta mortalidad en la infancia. El diagnóstico se basa en la evaluación de la actividad de la LAL y la identificación de mutaciones en el gen LIPA.
LÍPIDOS en 3 MINUTOS!!! (Fácil y Rápido)
Otras Enfermedades Lisosomales y Metabolismo Lipídico
Existen diversas enfermedades de almacenamiento lisosomal causadas por la deficiencia de otras hidrolasas ácidas, que afectan el metabolismo de diferentes macromoléculas y pueden tener repercusiones indirectas en el metabolismo lipídico o presentarse con alteraciones lipídicas:
- Esfingolipidosis: Enfermedades como la enfermedad de Gaucher (deficiencia de beta-glucocerebrosidasa), Tay-Sachs (deficiencia de hexosaminidasa A) y Niemann-Pick (deficiencia de esfingomielinasa) implican la acumulación de esfingolípidos.
- Mucopolisacaridosis: Causadas por la deficiencia de enzimas que degradan glicosaminoglicanos (antes mucopolisacáridos), como en la enfermedad de Hurler (deficiencia de alfa-L-iduronidasa) y el síndrome de Hunter (deficiencia de iduronato-2-sulfatasa).
- Glucogenosis tipo II (enfermedad de Pompe): Defecto en la alfa-glucosidasa ácida lisosomal, que lleva a la acumulación de glucógeno en los lisosomas.
Si bien estas enfermedades no afectan directamente la vía principal de degradación de triglicéridos y colesterol como lo hace la deficiencia de LAL, la disrupción general del metabolismo lisosomal puede tener consecuencias sistémicas, incluyendo alteraciones en la homeostasis lipídica.

tags: #metabolismo #lipidos #lisosoma