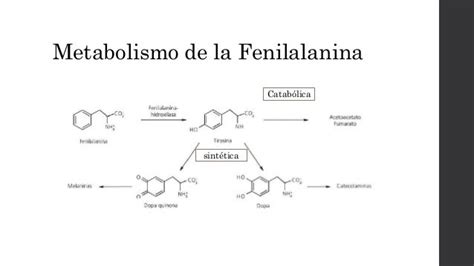
La fenilcetonuria (PKU) es una enfermedad genética causada por la deficiencia o ausencia de la enzima fenilalanina hidroxilasa (FAH). Esta enzima es crucial para metabolizar la fenilalanina (FA) en tirosina (TIR). Sin una FAH funcional, la fenilalanina se acumula en la sangre y el cerebro, provocando un daño progresivo en el sistema nervioso central. Si no se diagnostica y trata a tiempo, esta acumulación puede resultar en discapacidad intelectual severa.
Las hiperfenilalaninemias (HFA) son un grupo de trastornos metabólicos que incluyen la fenilcetonuria. Se clasifican según el nivel de FA plasmática, la tolerancia a la ingesta de este aminoácido y la actividad enzimática residual:
- Fenilcetonuria (PKU) clásica: Niveles de FA plasmática superiores a 20 mg/dL, TIR menor de 0.8 mg/dL, presencia de fenilquetonas en orina y actividad de FAH inferior al 1%. Estos pacientes tienen una tolerancia a la FA dietaria menor de 20 mg/kg/día.
- PKU moderada: Niveles de FA en plasma de 4 a 19 mg/dL, TIR en rango normal, actividad de FAH del 3% al 50%, y tolerancia a la FA de 20 a 25 mg de FA/kg/día.
- PKU leve: Niveles séricos de FA de 4 a 10 mg/dL y tolerancia a ingestas de FA mayores de 50 mg/kg/día.
- HFA leve: Niveles de FA plasmática de 4 a 10 mg/dL, nivel de TIR normal, actividad de FAH mayor del 50%, normalizándose después de los 6 meses. Estos casos generalmente no requieren dieta especial.
Adicionalmente, un pequeño porcentaje (1% a 2%) de las HFA se deben a defectos en el sistema cofactor de la FAH, siendo el más común el déficit de dehidropteridina reductasa (DHPR).
Clínica y Diagnóstico
Los síntomas de la fenilcetonuria, si no se trata, se manifiestan generalmente en los primeros meses de vida. La acumulación de fenilalanina en el cerebro interfiere con el transporte de otros aminoácidos aromáticos, dibásicos y neutros a través de las membranas celulares, incluida la barrera hematoencefálica. Esto reduce las concentraciones de aminoácidos intraneuronales, inhibe la hidroxilación de tirosina y triptófano, disminuye la síntesis proteica, afecta la proliferación dendrítica y la mielinización, y reduce la síntesis de serotonina, dopamina y norepinefrina.
Los signos y síntomas pueden incluir:
- Falta de interés por el entorno.
- Convulsiones.
- Eczema rebelde al tratamiento.
- Cabello, ojos y piel más claros que los de sus progenitores.
- Olor a humedad (a humedad o ratón) en el aliento, la piel y la orina, producido por la excreción de ácido fenilacético.
- Retraso en el desarrollo psicomotor (evidente alrededor de los 6 meses).
- En niños mayores: graves trastornos de conducta como agresividad, hiperactividad, rabietas, conductas autodestructivas y actitudes autistas.
- En adultos: regresión intelectual y deterioro neurológico asociado a desmielinización.
El diagnóstico precoz es fundamental para prevenir el daño neurológico. Los programas de detección neonatal, iniciados en la década de 1960 con la prueba de Guthrie, han permitido eliminar la fenilcetonuria como causa de retardo mental en muchos países. La incidencia global estimada es de aproximadamente 1:10.000 recién nacidos vivos (RNV), aunque varía significativamente entre grupos étnicos y regiones geográficas.
En Chile, la incidencia de PKU clásica se ha estimado en 1:21.509 RNV, y la de HFA en 1:14.416 RNV. Se han descrito más de 500 mutaciones en el gen de la FAH, y existe una correlación entre genotipo y fenotipo en aproximadamente el 79% de los casos.
Estudios prospectivos en niños con PKU detectados precozmente que abandonaron la dieta han demostrado la reversión de síntomas como disminución del coeficiente intelectual, cambios de conducta y déficit atencional al reintroducir la dieta restringida. Esto subraya la importancia de mantener la dieta de por vida.
Tratamiento de la Fenilcetonuria
El pilar fundamental del tratamiento de la fenilcetonuria es una dieta restringida en fenilalanina, que debe iniciarse lo antes posible, idealmente en el período neonatal, para prevenir el retardo mental y otras secuelas neurológicas.
Dieta Restringida en Fenilalanina
El objetivo principal de la dietoterapia es mantener los niveles de FA en sangre entre 2 y 10 mg/dL (120 - 600 uM/L), lo cual permite un crecimiento y desarrollo normal en los niños con PKU. El Consejo de Investigaciones Médicas (Medical Research Council) recomienda iniciar una dieta restringida en FA si el nivel de FA supera los 10 mg/dL con una dieta normal.
La dieta implica:
- Restricción de alimentos de origen animal: Carne, pollo, pescado, entre otros, debido a su alto contenido de fenilalanina.
- Restricción de cereales, frutas y verduras en cantidades variables según la tolerancia individual.
- Uso de un sustituto lácteo especial sin FA: Esencial para proporcionar proteínas de alto valor biológico y otros nutrientes necesarios.
- Fórmulas especiales bajas en proteínas: Se han desarrollado alimentos como fideos, galletas, pan y chocolates con bajo contenido proteico para aportar calorías adicionales, lograr saciedad y evitar transgresiones a la dieta.
En la primera semana de tratamiento, se suspende la lactancia materna y se administra el 100% del líquido requerido como fórmula especial sin FA (150ml/kg/día). El aporte calórico se completa con maltodextrina y aceite de soya, ajustado según sexo, edad y peso. Los niveles de FA en sangre se miden semanalmente mediante el método de inhibición bacteriana de Guthrie, y se ajusta la ingesta de FA en consecuencia.
El Instituto de Nutrición y Tecnología de los Alimentos (INTA) de la Universidad de Chile ha incorporado la lactancia materna como parte del tratamiento en PKU, observando que hasta el cuarto mes, la leche materna aporta el 50% de las proteínas, calorías y FA, manteniendo los niveles de FA en sangre por debajo de 6.0 mg/dL.
Suplementación Nutricional
La dieta restringida por sí sola no aporta adecuadamente nutrientes esenciales como zinc, selenio, hierro, cobre, cromo, vitamina B12 y calcio. Por ello, es necesaria su suplementación farmacológica o a través de la fórmula especial sin FA. También se presta atención a los ácidos grasos esenciales (araquidónico y docosahexaenoico), recomendando una razón entre linoleico y alfa linolénico de 1:15 para fomentar su síntesis.
Se ha observado que niños con PKU en dieta estricta pueden presentar desmineralización ósea, aumentando el riesgo de fracturas. La deficiencia de tirosina, un aminoácido esencial, puede disminuir la síntesis de dopamina y noradrenalina. Por ello, se sugiere suplementar con TIR (100-300 mg/kg/día) para mantener su nivel en sangre sobre 0.8 mg/dl.
Tratamiento Farmacológico
La sapropterina (Kuvan®), una forma sintética de tetrahidrobiopterina (BH4), ha sido aprobada para el tratamiento de la fenilcetonuria. Este medicamento puede ayudar a algunas personas a reducir los niveles de fenilalanina en sangre cuando se usa en combinación con la dieta, aunque no todas las personas responden a este tratamiento.
La pegvaliase-pqpz (Palynziq®) es una nueva terapia enzimática aprobada para adultos con fenilcetonuria cuyos niveles de fenilalanina no se reducen eficazmente con las terapias actuales.
Manejo en Situaciones Especiales
- PKU Materna: La dieta debe ser extremadamente estricta antes de la concepción y durante todo el embarazo para prevenir el efecto teratogénico de la HFA sobre el feto. Se recomienda mantener los niveles de FA por debajo de 4 mg/dL y el TIR plasmático sobre 0.8 mg/dl.
- Períodos de Infección, Ayuno o Cirugía: El aumento de FA plasmática en estas situaciones requiere disminuir la ingesta de FA en un 25%, aumentar el volumen de fórmula sin FA y potenciar la ingesta de energía para restablecer el balance metabólico.
Dieta equilibrada en PKU | Dra. Amaya Belanguer
Estilo de Vida y Apoyo
Vivir con fenilcetonuria implica un compromiso de por vida con la dieta y el seguimiento médico. Es fundamental:
- Educación y Concienciación: Informarse sobre la enfermedad y sus implicaciones.
- Seguimiento Médico Regular: Realizar análisis de sangre periódicos para controlar los niveles de fenilalanina, a menudo semanalmente durante el primer año y luego mensualmente.
- Planificación Alimentaria: Trabajar con un dietista registrado para crear planes de comidas individualizados, creativos y nutritivos.
- Uso de Herramientas de Medición: Utilizar tazas, cucharas medidoras y balanzas de cocina para asegurar la precisión en las porciones.
- Evitar el Aspartamo: Este edulcorante artificial contiene fenilalanina y debe ser evitado.
- Apoyo Psicosocial: Unirse a grupos de apoyo y buscar ayuda de familiares y amigos puede ser de gran utilidad.
- Adaptación Familiar: Crear un ambiente donde las comidas sean momentos familiares, enfocándose en las interacciones y no solo en los alimentos.
- Planificación para Eventos Especiales: Preparar comidas o refrigerios para salidas, fiestas o viajes.
- Colaboración Escolar: Informar a maestros y personal escolar sobre las necesidades dietéticas del niño.
El pronóstico para las personas con fenilcetonuria es muy alentador si la dieta se sigue estrictamente desde el nacimiento, permitiendo un desarrollo neurológico normal y una vida plena.

tags: #desordenes #del #metabolismo #fecilcetonuria #tratamiento