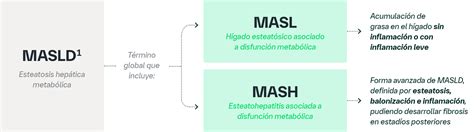
La enfermedad hepática esteatósica asociada a disfunción metabólica (MASLD), anteriormente conocida como enfermedad del hígado graso no alcohólico (NAFLD), se caracteriza por la acumulación excesiva de lípidos en los hepatocitos.
MASLD: Definición y Terminología
La enfermedad hepática esteatósica asociada con disfunción metabólica se define como la presencia de esteatosis hepática en individuos con al menos un factor de riesgo metabólico, como obesidad o dislipidemia, y un consumo mínimo o nulo de alcohol. El cambio en la terminología de NAFLD a MASLD y de esteatohepatitis no alcohólica (NASH) a esteatohepatitis asociada a disfunción metabólica (MASH) refleja mejor la vinculación de estos trastornos con el síndrome metabólico y factores de riesgo metabólicos, no solo la obesidad.
La esteatohepatitis asociada a disfunción metabólica (MASH) es un subconjunto histológicamente definido de MASLD que se caracteriza por la presencia de esteatosis hepática con daño inflamatorio de los hepatocitos. La diferenciación entre MASLD y MASH es difícil sin biopsia hepática, y el aumento de las enzimas hepáticas no es un predictor sensible para identificar MASH. La presencia de MASH confiere un mayor riesgo de progresión a fibrosis avanzada o cirrosis.

Patogenia de la MASLD y MASH
La patogenia de la MASLD y MASH es compleja, pero la resistencia a la insulina se considera un factor clave. Esta resistencia aumenta la liberación de ácidos grasos libres en el hígado y la lipogénesis hepática de novo. Otros mecanismos potenciales incluyen la reducción de la síntesis de lipoproteínas de muy baja densidad (VLDL), el aumento de la síntesis de triglicéridos hepáticos, factores genéticos relacionados con el control de lípidos y posibles alteraciones en la producción de ácido úrico hepático.
La inflamación continua puede estimular las células estrelladas hepáticas, lo que conduce a la fibrosis. Si la fibrosis es avanzada, la MASH puede causar cirrosis (a veces con progresión a carcinoma hepatocelular) y hipertensión portal. Sin embargo, la MASH también puede regresar a una forma de esteatosis simple.
Epidemiología y Factores de Riesgo
La MASLD se diagnostica con mayor frecuencia en pacientes entre 50 y 60 años, pero su incidencia está aumentando en grupos de edad más jóvenes, incluyendo adolescentes, debido a la epidemia de obesidad. Los pacientes afectados suelen presentar al menos un factor de riesgo cardiometabólico, como:
- Obesidad (IMC > 25 kg/m², o > 23 en asiáticos) o circunferencia de la cintura aumentada.
- Diabetes mellitus tipo 2, intolerancia a la glucosa o niveles elevados de glucosa en ayunas (HbA1c > 5,7%).
- Hipertensión arterial (presión arterial > 130/85 mm Hg o en tratamiento antihipertensivo).
- Dislipidemia (triglicéridos > 150 mg/dL, o en tratamiento hipolipemiante; o colesterol HDL bajo).
- Síndrome metabólico.

Signos y Síntomas
La mayoría de los pacientes con MASLD son asintomáticos, especialmente aquellos con esteatosis simple. Sin embargo, algunos pueden experimentar:
- Astenia (fatiga).
- Malestar general.
- Dolor en el cuadrante superior derecho del abdomen.
En aproximadamente el 75% de los pacientes con MASH, se desarrolla hepatomegalia (aumento del tamaño del hígado). En casos de fibrosis hepática avanzada, puede presentarse esplenomegalia, que a menudo es el primer indicio de hipertensión portal. Los pacientes con cirrosis secundaria a MASH pueden no mostrar los síntomas habituales de la hepatopatía crónica. En etapas avanzadas, puede haber ictericia (coloración amarillenta de piel y ojos) debido a la acumulación de bilirrubina.
Diagnóstico de MASLD y MASH
El diagnóstico de MASLD debe sospecharse en pacientes con síndrome metabólico o factores de riesgo metabólicos, antecedentes limitados de ingesta de alcohol, y anomalías de laboratorio inexplicables que sugieren hepatopatía.
El diagnóstico formal de MASLD requiere evidencia de:
- Esteatosis hepática (detectada por estudios de imagen, biomarcadores o biopsia).
- Al menos 1 factor de riesgo cardiometabólico.
- Antecedentes de consumo de alcohol moderado (no más de 2 bebidas al día en mujeres o 3 en hombres).
- Serología negativa para hepatitis B y C.
El diagnóstico de MASH requiere evidencia de inflamación hepática y esteatosis, y generalmente necesita una biopsia hepática que muestre inflamación y lesión hepatocelular, con o sin fibrosis. La biopsia revela infiltración grasa macrovesicular en más del 5% de los hepatocitos, hepatocitos dañados ("globosos") e inflamación crónica.
Los estudios de diagnóstico por imágenes como ecografía, TC y RM pueden identificar la esteatosis hepática. Las medidas no invasivas de la fibrosis, como la elastografía transitoria, elastografía por ecografía o elastografía por RM, son valiosas para evaluar la gravedad de la esteatosis y estimar la fibrosis, evitando en muchos casos la necesidad de biopsia. Sin embargo, estas pruebas no detectan la inflamación característica de la MASH y no pueden diferenciarla de otras causas de esteatosis hepática.
Tratamiento de MASLD y MASH
La piedra angular del tratamiento para la mayoría de los pacientes con MASLD es la eliminación de las causas y el control de los factores de riesgo metabólicos. Esto incluye:
- Pérdida de peso: Una reducción del 7-10% del peso corporal puede disminuir la grasa hepática y frenar la progresión de la fibrosis.
- Tratamiento de la dislipidemia y la hiperglucemia.
- Suspensión de fármacos o toxinas que puedan contribuir a la enfermedad.
Las intervenciones como la pérdida de peso han demostrado reducir la inflamación hepática, la esteatosis y la fibrosis.
En pacientes seleccionados con fibrosis en estadio F2 a F3 (moderada a grave), se puede utilizar el medicamento resmetirom, un agonista del receptor beta tiroideo. En ensayos clínicos, el resmetirom ha demostrado revertir la esteatosis y la MASH, y mejorar la fibrosis. Se recomienda monitorizar las pruebas hepáticas antes y durante el tratamiento.
Otras terapias emergentes para la MASLD actúan sobre diversas vías moleculares, como los receptores activados por proliferadores de peroxisomas alfa (PPAR-alfa) y los moduladores del péptido similar al glucagón (GLP).
Esteatosis Hepática | NASH / MASLD
Miopatías Metabólicas: Definición y Causas
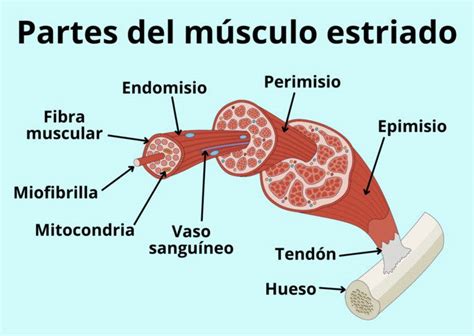
Las miopatías metabólicas son un grupo de trastornos genéticos raros que afectan el músculo esquelético, interfiriendo con los procesos celulares que generan la energía necesaria para la contracción muscular. Los defectos genéticos causan deficiencias enzimáticas que afectan el metabolismo del glicógeno, los lípidos, la cadena respiratoria mitocondrial o las purinas.
Los músculos necesitan trifosfato de adenosina (ATP) para funcionar, el cual se produce a partir de azúcares y grasas a través de complejas vías químicas. La falta de ciertas enzimas clave en estas vías conduce a las miopatías metabólicas.
Clasificación de las Miopatías Metabólicas
Las miopatías metabólicas se agrupan etiopatogénicamente en:- Trastornos del metabolismo del glicógeno (Glicogenosis): Afectan la síntesis o degradación del glicógeno. Las más comunes que afectan al músculo son la enfermedad de Pompe (déficit de maltasa ácida) y la enfermedad de McArdle (déficit de miofosforilasa).
- Trastornos del metabolismo de los lípidos.
- Defectos de la cadena respiratoria mitocondrial.
- Trastornos del metabolismo de purinas (deficiencia de mioadenilato deaminasa).
Enfermedad de McArdle (Glicogenosis Tipo V)
Se manifiesta por intolerancia al ejercicio, con cansancio, fatigabilidad, mialgias, calambres y debilidad, especialmente con ejercicio isométrico breve e intenso. Alrededor del 50% de los pacientes experimentan mioglobinuria. Un fenómeno característico es el "segundo aire", donde los síntomas mejoran al continuar con ejercicio de menor intensidad.
El diagnóstico se basa en la clínica, pruebas de ejercicio isquémico (curva plana de lactato), y biopsia muscular que muestra ausencia de fosforilasa muscular. El tratamiento no es curativo e incluye evitar el ejercicio intenso, una dieta alta en carbohidratos complejos y suplementación con vitamina B6 en algunos casos.
Enfermedad de Pompe (Glicogenosis Tipo II)
Causada por el déficit de la enzima α-glucosidasa ácida, lleva a la acumulación de glicógeno en los lisosomas, especialmente en músculo esquelético, corazón e hígado. Existe un espectro clínico continuo:
- Forma infantil precoz: Hipotonía, debilidad generalizada, miocardiopatía severa, insuficiencia respiratoria.
- Forma juvenil: Debilidad muscular progresiva de predominio proximal, dificultad respiratoria, especialmente en decúbito.
- Forma de inicio tardío: Debilidad lentamente progresiva, predominio proximal y axial, debilidad diafragmática desproporcionada, disnea.
El diagnóstico se basa en la clínica, niveles elevados de creatinkinasa (CK), estudios cardiológicos y de función pulmonar. El tratamiento incluye terapia de reemplazo enzimático (ERT) para las formas infantiles y juveniles, y manejo de síntomas respiratorios y musculares para las formas de inicio tardío.
Signos y Síntomas de Miopatías Metabólicas
Los síntomas de las miopatías metabólicas varían según el tipo específico y la gravedad, pero comúnmente incluyen:
- Debilidad muscular: Puede ser progresiva o intermitente.
- Cansancio o fatiga después del ejercicio o actividad física.
- Dolor muscular (mialgias) después del esfuerzo.
- Músculos hinchados o sensibles.
- Mioglobinuria (presencia de mioglobina en la orina), especialmente en la enfermedad de McArdle, indicando daño muscular agudo (rabdomiólisis).
- En algunos casos, intolerancia al ejercicio, calambres, debilidad de cintura pélvica, dificultad para subir escaleras, o problemas respiratorios.
Algunas personas con miopatías metabólicas pueden no presentar síntomas evidentes, o tener síntomas leves.
Diagnóstico de Miopatías Metabólicas
El diagnóstico de miopatías metabólicas se basa en:
- Historia clínica detallada: Incluyendo síntomas, desencadenantes potenciales, ejercicio, historial familiar de consanguinidad o síntomas relacionados.
- Examen físico: Búsqueda de debilidad muscular (generalizada o localizada), hipertrofia muscular, o anormalidades en la relajación muscular.
- Pruebas de laboratorio: Creatinkinasa (CK) elevada es común, aunque los niveles pueden ser normales. Se buscan marcadores de rabdomiólisis.
- Pruebas de ejercicio: Como la prueba de ejercicio isquémico para la enfermedad de McArdle.
- Biopsia muscular: Para examinar el tejido muscular en busca de anomalías específicas, como acumulación de glicógeno o deficiencias enzimáticas.
- Estudios genéticos moleculares: Para identificar mutaciones específicas en los genes asociados con las miopatías metabólicas.
Tratamiento de Miopatías Metabólicas
El tratamiento de las miopatías metabólicas varía significativamente según la enfermedad específica y se enfoca en controlar los síntomas y minimizar la progresión de la debilidad muscular. Las estrategias comunes incluyen:
- Cambios en la dieta: Adecuados aportes de glucosa, especialmente en situaciones de sobreexigencia metabólica. Dietas altas en carbohidratos complejos y bajas en grasa pueden ser útiles en la enfermedad de McArdle.
- Modificación de la actividad física: Evitar situaciones que precipiten síntomas (ejercicio intenso) y adaptar el nivel de actividad.
- Terapia física y ocupacional: Para mantener la fuerza muscular, la movilidad y la independencia funcional.
- Suplementos: Como vitamina B6 en la enfermedad de McArdle, o coenzima Q10 en algunos trastornos mitocondriales.
- Manejo de complicaciones: Como la insuficiencia respiratoria o renal aguda asociada a rabdomiólisis masiva.
- Terapias específicas: Como la terapia de reemplazo enzimático (ERT) en la enfermedad de Pompe.
El objetivo principal es mejorar la calidad de vida y mantener la autonomía del paciente.
Esteatosis Hepática | NASH / MASLD
tags: #miopatia #metabolica #hepatico