La deficiencia de carnitina es una condición que resulta de una ingesta inadecuada de este aminoácido o de la incapacidad del organismo para metabolizarlo. Esta deficiencia puede manifestarse a través de un grupo heterogéneo de trastornos, afectando principalmente el metabolismo muscular, lo que puede desencadenar miopatías, hipoglucemia y miocardiopatía. En niños, los síntomas pueden incluir encefalopatía hipoglucémica e hipocetósica. El tratamiento habitual consiste en la administración de L-carnitina en la dieta.
El aminoácido carnitina desempeña un papel crucial en el transporte de ésteres de ácidos grasos de cadena larga, como el acil-coenzima A (CoA), hacia el interior de las mitocondrias de los miocitos. En este compartimento celular, los ácidos grasos son oxidados para generar energía. La carnitina se obtiene tanto a través de la dieta, especialmente de alimentos de origen animal, como por síntesis endógena en el organismo.
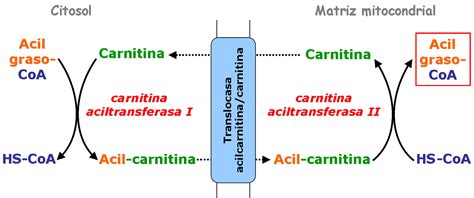
Causas de la Deficiencia de Carnitina
Existen diversas causas que pueden conducir a una deficiencia de carnitina:
- Ingestión inadecuada: Esto puede deberse a dietas restrictivas o de moda, falta de acceso a alimentos ricos en carnitina, o nutrición parenteral total prolongada.
- Incapacidad para metabolizar la carnitina: Deficiencias enzimáticas específicas, como la deficiencia de carnitina palmitoiltransferasa, metilmalonicaciduria, propionicacidemia e isovalericacidemia, impiden el correcto metabolismo de la carnitina.
- Disminución de la síntesis endógena: Un trastorno hepático grave puede afectar la capacidad del hígado para sintetizar carnitina.
- Pérdida excesiva de carnitina: Condiciones como diarrea crónica, diuresis excesiva o hemodiálisis pueden resultar en una pérdida significativa de carnitina.
- Trastorno hereditario renal: Ciertas condiciones genéticas provocan que la carnitina se escape de los túbulos renales, resultando en su excreción excesiva.
- Aumento de los requerimientos de carnitina: Situaciones como la cetosis, o una alta demanda para la oxidación de lípidos durante enfermedades graves (sepsis, quemaduras extensas) o después de cirugías del tracto gastrointestinal, incrementan la necesidad de carnitina.
- Alteraciones mitocondriales: El uso de ciertos medicamentos, como la zidovudina, o la presencia de alteraciones mitocondriales pueden disminuir los niveles de carnitina en el músculo.
- Uso de ácido valproico: Este medicamento también se ha asociado con la disminución de los niveles de carnitina.
La deficiencia de carnitina puede presentarse de forma generalizada (sistémica) o afectar predominantemente a los músculos (miopática).
Síntomas de la Deficiencia de Carnitina
Los síntomas de la deficiencia de carnitina y la edad de aparición son variables y dependen de la causa subyacente. Las manifestaciones clínicas pueden incluir:
- Necrosis muscular
- Mioglobinuria
- Miopatía por depósito de lípidos
- Hipoglucemia
- Hígado graso
- Hiperamoniemia
- Dolores musculares
- Fatiga
- Confusión
- Miocardiopatía
Diagnóstico de la Deficiencia de Carnitina
El diagnóstico de la deficiencia de carnitina se basa en pruebas específicas que varían según la edad del paciente:
- En recién nacidos: La espectrometría de masa es una prueba de detección en sangre utilizada para diagnosticar la deficiencia de carnitina palmitoiltransferasa. El diagnóstico prenatal es posible mediante el análisis de células de las vellosidades amnióticas.
- En adultos: El diagnóstico definitivo se establece midiendo los niveles de acilcarnitina en suero, orina y tejidos. Para la deficiencia sistémica, se analizan músculo e hígado, mientras que para la deficiencia miopática, solo se analiza el músculo.
En el contexto de enfermedades metabólicas congénitas, los pacientes pueden acudir a los servicios de urgencias pediátricas por diversos motivos, algunos relacionados con su patología de base y otros no. Un estudio retrospectivo analizó 107 visitas de pacientes con enfermedades metabólicas congénitas a un hospital de tercer nivel, identificando los procesos respiratorios como el motivo de consulta más frecuente (30,8%). Las visitas por vómitos en pacientes con trastornos relacionados con proteínas fueron más rápidas en acudir a urgencias. Aproximadamente un tercio de las visitas resultaron en ingreso hospitalario, la mitad de ellas debido a descompensación metabólica.
Tratamiento de la Deficiencia de Carnitina
El tratamiento de la deficiencia de carnitina se enfoca en evitar situaciones que puedan precipitar una descompensación y en la suplementación con L-carnitina:
- Evitar el ayuno y la actividad física extenuante: Estas medidas son fundamentales para prevenir episodios de hipoglucemia.
- Intervenciones dietéticas: El consumo de maicena sin cocinar antes de acostarse puede ayudar a prevenir la hipoglucemia nocturna.
- Suplementación con L-carnitina: En casos de deficiencia por ingestión inadecuada, aumento de requerimientos, pérdida excesiva, disminución de la síntesis o ciertas deficiencias enzimáticas, se administra L-carnitina, generalmente en dosis de 25 mg/kg por vía oral cada 6 horas. En el caso de la deficiencia primaria de carnitina, las dosis de levocarnitina oral pueden ser de 100-400 mg/kg al día, distribuidas en tres tomas.
- Suplementos adicionales: Algunos pacientes pueden requerir suplementos con triglicéridos de cadena mediana y ácidos grasos esenciales, como el ácido linoleico y linolénico.
La prevalencia exacta de la deficiencia de carnitina varía entre etnias, estimándose entre 1/20.000 y 1/70.000 nacimientos en Europa y EE. UU., y 1/40.000 en Japón. La enfermedad suele manifestarse en la infancia, entre los 3 meses y los 2 años, con síntomas como hipoglucemia hipocetósica, problemas de alimentación, irritabilidad, letargia y hepatomegalia, a menudo desencadenados por estrés metabólico. En aproximadamente la mitad de los casos, se presenta hipotonía muscular y miocardiopatía progresiva. La presentación en adultos puede ser más sutil, con fatiga y disminución de la resistencia, aunque también se han descrito miocardiopatía dilatada, arritmias e incluso muerte súbita cardíaca. En algunos casos, los adultos pueden ser asintomáticos.
La enfermedad está causada por mutaciones en el gen SLC22A5, que codifica para el transportador de carnitina OCTN2. Este transportador es esencial para el ingreso de L-carnitina en las células. El diagnóstico se basa en la detección de concentraciones muy bajas de carnitina en plasma y se confirma mediante análisis de transporte de carnitina en fibroblastos o la identificación de mutaciones bialélicas en el gen SLC22A5. La miopatía lipídica, la acumulación de lípidos en el músculo e hígado, y la pérdida renal de carnitina son hallazgos característicos.
Suplementación Con L-Carnitina - Mitos, Efectos Reales Y Cómo Tomarla
El tratamiento estándar para la deficiencia de carnitina es la terapia con levocarnitina. Si bien los avances en el diagnóstico y tratamiento han mejorado la supervivencia y calidad de vida de los pacientes con enfermedades metabólicas congénitas, es crucial el manejo adecuado de las descompensaciones metabólicas agudas, las cuales pueden requerir ingreso hospitalario. La falta de ensayos clínicos de alta calidad metodológica sobre la suplementación con carnitina subraya la importancia de basar las decisiones clínicas en la experiencia, las circunstancias individuales y la preferencia del paciente, mientras se continúa la vigilancia y se promueven futuras investigaciones.
tags: #carnitina #en #defectos #congenitos #del #metabolismo